Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Krishna Singh and Version 2 by Rita Xu.
Germline mutations in Breast cancer susceptibility genes 1 and 2 (BRCA1 and BRCA2) cause breast, ovarian, and other cancers, and the chemotherapeutic drug doxorubicin (Dox) is widely used to treat these cancers. Dox use is limited by the latent induction of severe cardiotoxicity known as Dox-induced cardiomyopathy, for which there are no specific treatments currently available. Dox is administered into the systemic circulation, where it readily translocates into sub-cellular compartments and disrupts the integrity of DNA.
- BRCA2
- BRCA1
- doxorubicin
- breast cancer
1. Dox-Induced Cardiotoxicity (DIC)
Dox-induced cardiotoxicity (DIC) is notably common in patients receiving Dox treatment and typically manifests as cardiomyocyte injuries subjected to the mechanisms of Dox that were meant for neoplastic cells. Clinically, DIC is either acute or chronic, depending on the dosing regimen, age, and cardiovascular health of the patients. The recent incidence rate of acute DIC is ~30% and typically detected 2–3 days after Dox administration, with ~50% mortality after 1-year diagnosis. Acute DIC is reversible and often manifests with tachycardia and ventricular premature beats shortly following administration. Histological features of acute DIC in the myocardium include interruption of myofibrils, cytoplasmic vacuolation, and sparsity in cardiomyocytes [1][11]. The incidence of chronic DIC is ~2–20% and typically manifests as a complication of breaching Dox dosage tolerance after weeks or months of administration, featuring irreversible cardiomyopathy, prominent left ventricular enlargement, and heart failure [2][3][4][10,12,13]. The mechanisms of DIC are summarized in Figure 1.
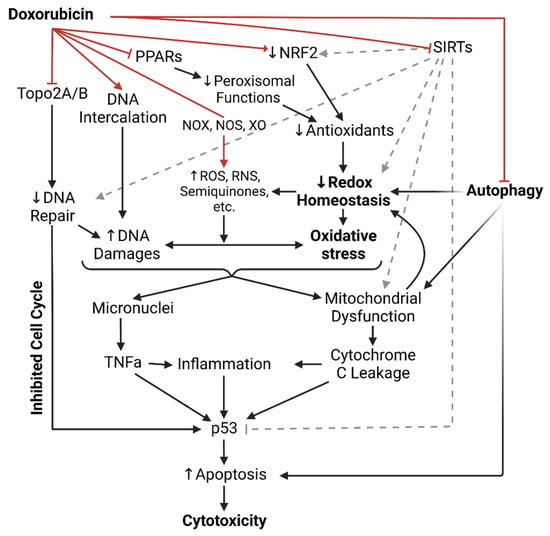
Figure 1. Illustrated summary of all doxorubicin’s mechanisms of cytotoxicity up to date. Red arrows indicate direct actions of doxorubicin. ROS = Reactive oxygen species; RNS = Reactive nitrogen species; NOX = NADPH oxidase; NOS = Nitric oxide synthase; XO = Xanthine oxidase; PPAR = Peroxisome proliferator-activated receptors; NRF2 = Nuclear factor erythroid 2-related factor 2; SIRTs = Sirtuin-like proteins; Topo2A/B = Topoisomerase 2A/B; TNFa = Tumor necrosis factor alpha.
2. Intercalation of DNA and Topoisomerase II Inhibition
It has been generally established that free radical-associated DNA damage is more likely to occur when Dox concentration breaches the tolerance dosage [5][14]. Instead, Dox-associated cytotoxic impact on the cell’s genome is often attributed to DNA intercalation and Dox-dependent topoisomerase II inhibition [6][7][15,16]. Notably, considering the highly oxidative environment subjected to cardiac tissues by default, both mechanisms likely damage genomic integrity to a similar degree of significance in DIC.
The drug’s potent mechanism is DNA intercalation, achieved via its anthraquinone ring. Dox inserts itself between DNA bases and stabilizes the interaction through hydrogen bonding, facilitated by its hydroxyl and daunosamine sugar’s amino groups. Dox-DNA complexes induce torsional stress in the DNAs superhelical structures, leading to double-strand breaks and apoptosis. In addition, Dox can bind and inhibit DNA and RNA polymerases, disrupting replication, transcription, and DNA repair [7][16].
An additional Dox-associated cytotoxic impact on the cell’s genome is topoisomerase II inhibition [6][15]. Topoisomerase II (TopoII) is a well-characterized enzyme that functions in replication (TopoIIa) or DNA repair and gene transcription (TopoIIb). Typically, it is recruited to associate with the DNA strand and induce coordinated DNA double-strand breaks as a response to alleviate torsional stress, relax positive supercoils, and unlink intertwined strands, in turn stabilizing the DNAs superhelical state. TopoII inhibition is also Dox’s mechanism designed for rapidly proliferating cells. Dox binds and inhibits both TopoIIa and IIb during their actions, forming Dox-TopoII-DNA complexes that induce unaccounted-for double-strand breaks, hindering replication and gene expression, and resulting in cell death [6][15]. Indeed, Dox efficacy is dependent on TopoII concentration in the cells [8][2]. Dox-TopoII-DNA complexes can be reversed by the dissociation of Dox upon turnover, suggesting that Dox exposure duration is a key factor in Dox cytotoxicity [9][17].
The genomic impacts of Dox are typically observed at 0.01–5 μM Dox concentrations, followed by p53 upregulation [8][2]. p53 is constitutively expressed and preferably suppressed in cells. Its activation regulatory competes with various cell growth and DNA repair pathways; hindrance in the cell’s growth/survival (e.g., downregulated E2F, the growth transcription factor) and/or improper DNA repair promote p53 activation [10][11][18,19]. Activated p53 facilitates cell cycle arrest and programmed cell death [12][20]. While Dox is designed to tarnish cellular replication and prompt p53’s pro-apoptotic action in the neoplastic cell population, in DIC, Dox-treated cardiac tissues demonstrate impaired vital gene expression, DNA damage, and dysregulated p53, among other complications [13][14][21,22]. In this notion, genetic and pharmacologic inhibition of p53 have been shown to attenuate acute DIC [15][16][17][18][19][23,24,25,26,27]; however, disrupted p53 activity has been associated with exacerbated cardiac dysfunction in animal models [14][22] and is also a feature of Dox-resistant tumor cells [20][28], suggesting a selectivity challenge for p53 targeting therapeutic strategies.
Moreover, in DIC, while cardiomyocytes are non-dividing cells, they preferentially express TopoIIb over TopoIIa. This expression is adopted by both the cardiomyocytes’ genomes and mitochondria. A single cardiomyocyte can have between 5000 and 8000 mitochondria, and mitochondria are vital for cardiac function, regulating essential lipid oxidation and redox balance [21][29]. Therefore, Dox’s inhibiting TopoIIb and the formation of Dox-TopoIIb-DNA complexes in cardiomyocytes promote DNA damage and oxidative stress as a feature of DIC.
3. NADPH Oxidases, Nitric Oxide Synthases, and Xanthine Oxidase
DICs oxidative stress has been well characterized by three enzymes: NADPH oxidases (NOXs), nitric oxide synthases (NOSs), and xanthine oxidases (XOs), which were previously mentioned as metabolizers of Dox into semiquinone radicals. In addition to metabolizing Dox into radical intermediates, these enzymes pathologically contribute to oxidative stress by facilitating the production of oxidative radicals [22][23][24][30,31,32]. However, targeting these enzymes clinically remains under consideration due to conflicting observations attributed to their physiological oxidative significance. For instance, as XO mediates oxygen radical generation and exacerbates DIC in Dox-treated mice [25][33], febuxostat, an inhibitor of XO that reduces ROS production in the myocardium, has yet to be used clinically [26][27][34,35]. The NOXs enzymes are significant sources of superoxide production, with NOX2 and NOX4 being the primary contributors to Dox-induced oxidative stress [28][36]. The antioxidant treatments irisin and osteocrin inhibit NOX2 and NOX4, respectively, and result in the attenuation of Dox-induced oxidative stress in the heart [29][30][37,38]. However, NOXs inhibitors have yet to be considered clinically as their oxidative roles have physiological significance [31][39]. NOX2-deficient mice on high-fat diets developed severe glucose metabolism disorders, suggesting that the NOX enzymes may be a sensitive target to modulate Dox cardiotoxicity [32][40].
Nitric oxide (NO) derived from NOS enzymes can interact with concurrent radicals to produce more radicals, such as the peroxynitrite anion (ONOO-). Indeed, available antioxidant treatments for Dox-induced cardiotoxicity involve altering NOSs expression, such as levosimendan [33][41] and vitamin C [34][42]. Dox-treated cardiomyocytes show increased inducible NOS (iNOS) expression, and iNOS-deficient mice show attenuated Dox-induced generation of ONOO [35][43]. However, studies have also shown that the lack of NO bioavailability exacerbates Dox-induced cardiotoxicity [36][44]. Notably, endothelial NOS (eNOS) catalyzes the biological synthesis of NO, which is an essential regulator of endothelial function and vasotone [37][45]. Indeed, eNOS-deficient and overexpressing mice showed reduced and increased susceptibility to Dox-induced oxidative stress, respectively [38][46]. On the other hand, Dox has been shown to impair eNOS activation while facilitating ROS-mediated oxidative stress [39][47]. Moreover, eNOS-deficient female mice were shown to have aggravated Dox-induced oxidative stress and cellular damage [40][48].
4. Antioxidants in Dox-Induced Cardiotoxicity
Relatively, cardiac tissues engage in an overall highly oxidative metabolic environment to sustain their essential systemic function. Approximately 70% of the energy in the heart is derived from the oxidation of fatty acids within the mitochondria and peroxisomes, which rely heavily on lipid-trafficking mechanisms [41][49]. Naturally, cardiomyocytes express sensitive levels of endogenous antioxidant enzymes (e.g., superoxide dismutase, glutathione peroxidase, glutathione S-transferase, heme oxygenase-1, catalase, etc.) [42][50]. Tipping the redox balance, as in antioxidant deficiency, easily renders cardiac tissues vulnerable to oxidative stress. Indeed, Dox impairs the antioxidants’ bioavailability in cardiomyocytes, depending on the duration and dosage of Dox exposure [43][51]. Conversely, increasing antioxidants’ activity enhances the cells’ redox capacity, obstructs free radical-related injury to DNA, and attenuates DIC [44][45][52,53]. However, adjusting antioxidant bioavailability remains a challenge in the clinical setting of DIC. For instance, apigenin, which enhances antioxidants in cardiomyocytes, has yet to be considered for clinical practice [46][54]. Nrf2, a transcription factor regulating antioxidant enzymes, is downregulated in DIC, which is rescued by the yet-to-be clinically implemented irisin, a benefit additional to the drug’s inhibition of NOX2 that mitigates oxidative DIC [47][55].
5. Peroxisomes in Dox-Induced Cardiotoxicity
Cellular peroxisomes are organelles that maintain redox and lipid homeostasis via fatty acid β-oxidation and detoxification of metabolic byproducts, such as hydrogen peroxide (H2O2), polyamines, and glyoxylate, as well as several xenobiotics [48][56]. Notable peroxisomal enzymes include Catalase and Peroxidases, such as the Peroxiredoxins, that process various oxidative radicals into water and oxygen, thereby maintaining oxidative balance. Dysfunctional peroxisomes, as in developing Niemann–Pick type C disease [49][57], lead to radical accumulation, oxidative stress, and the downstream risk of multiple-organ failures. Indeed, aging cells, which become progressively peroxisomal inefficient, feature elevated ROS levels [50][58]. Conversely, overexpressing peroxiredoxin-1 attenuates oxidative stress and DIC in cardiomyocytes [51][52][59,60].
As oxidative stress and inflammation typically occur together, peroxisomal functions are closely linked to the inflammatory response. Likewise, chronic inflammation also features elevated ROS and oxidative cellular injuries. Moreover, peroxisomal enzymes also facilitate the degradation of pro-inflammatory mediators (e.g., prostaglandins, thromboxanes, leukotrienes, and prostacyclins) and the biogenesis of anti-inflammatory metabolites (e.g., omega-3 fatty acids) [53][61]. Notably, omega-3 fatty acids are precursors of potent anti-inflammatory factors such as resolvins, maresins, and protectins [54][62]; enhancing the conversion of omega-3 fatty acids to these molecules is a feature of non-steroidal anti-inflammatory drugs (NSAIDs), such as aspirin [55][63].
Mitigating DIC approaches from peroxisome functions have heavily focused on the peroxisome proliferator-activated receptors (PPARs). The PPARs are a family of nuclear transcription factors that, upon activation, upregulate peroxisome proliferators, which induce an increase in cellular peroxisome number, size, and functions [56][64]. In humans, three closely related PPAR subtypes have been identified. PPAR-δ is expressed ubiquitously and at higher levels than the other two. PPAR-γ is mainly found in adipose tissues and, to a lesser extent, immune cells (monocytes, macrophages, etc.). PPAR-α is rich in hepatocytes, cardiomyocytes, skeletal muscles, and other peripheral tissues with active lipid oxidation. Notably, of the three, PPAR-α exerts the highest affinity for lipids, regulating the escorts of unsaturated and saturated fatty acids via cytosolic fatty-acid binding proteins to the peroxisomes and mitochondria for lipid β-oxidation [57][65].
Dox inhibits PPARγ in the adipose tissues of mice, leading to the loss of the storage of blood glucose and lipid, thereby causing hyperglycemia and hyperlipidemia, which are high-risk factors for insulin resistance, atherogenesis, and cardiovascular diseases [58][66]. PPAR-α-null mice exhibit a loss of fatty acid oxidative capacity, leading to increased lipid accumulation, reduced ketone bodies, a lack of gluconeogenesis, and metabolic switching to fatty acid usage in the heart during starvation. These culminate in cardiac dysfunction, myocardial damage, and fibrosis [59][67]. The heart of tumor-bearing Dox-treated mice also showed inhibited PPARα, but such is absent in their tumors; conversely, the same paper demonstrates that fenofibrate (FENO) treatment, an agonist of PPARα, and overexpression of PPARα in these mice enhanced cardiac function and salvaged DIC without affecting tumor progression [60][68]. Notably, FENO was shown to attenuate DIC in mice by improving endothelial function and upregulating eNOS expression and activation via Akt [61][69]. Outside of modulating the PPARs, there are little to no studies addressing the role of peroxisomes more directly for clinical applications against DIC [52][60].
6. Sirtuins Deacetylate Dox-Induced Cardiotoxicity
The Sir2 and Sir2-like proteins, together referred to as sirtuins (SIRTs), are NAD-dependent deacetylase enzymes that make up the evolutionarily conserved class III histone-deacetylases in humans [62][70]. A distinct NAD/FAD-binding domain characterizes the class III HDACs. In humans, seven sirtuins have been reported and localized: the nucleus (SIRT1, SIRT2, SIRT3, SIRT6, SIRT7), cytoplasm (SIRT1, SIRT2), and mitochondria (SIRT3, SIRT4, SIRT5) [62][70].
The SIRTs have been reported to regulate aging, metabolism, inflammation, apoptosis, and maintaining redox balance in cardiac cells [63][71]. SIRT1 has been linked to cardiovascular diseases [64][72]. SIRT3, 6, and 7 regulate aging, apoptosis, and oxidative stress in cardiomyocytes and are linked to cardiac hypertrophy [65][66][67][73,74,75]. The roles of SIRT4 and 5 in the heart remain under-investigated.
SIRT3 deacetylase is expressed abundantly in cardiomyocytes and plays a crucial role in regulating mitochondrial function, proliferation, and maintenance of the mitochondrial genome, all of which are essential for cardiac metabolism. The mitochondria also engage in lipid oxidation, similar to peroxisomes, regulating the redox balance. Notably, ~20% of mitochondrial proteins are regulated via reversible lysine acetylation [68][76], and the mitochondria contain high levels of NAD and NADH [69][77]. Mitochondrial acyl-CoA dehydrogenase and synthase facilitate long-chain fatty acid oxidation and, thereby, lipid metabolism in cardiomyocytes [70][78]. SIRT3 regulates these enzymes and also those of the tricarboxylic acid cycle, electron transport chain subunits, and ATP synthase [71][79]. SIRT3 also regulates mitochondrial ROS formation via the antioxidants SOD2 [72][80] and Ku70, a factor of the non-homologous end-joining DNA repair pathway [73][81]. SIRT3 knock-down animals exhibit a high risk for cardiac hypertrophy, oxidative stress, diminished cardiac ATP, and increased mitochondrial fragmentation [74][75][76][82,83,84]. Dox-treated cardiomyocytes exhibit mitochondrial dysfunction that contributes to oxidative stress and impaired lipid metabolism, as featured in DIC [77][85]. Resveratrol, which activates SIRT3, in co-treatment with Dox, attenuates mitochondrial ROS production [78][79][86,87].
Cardiomyocytes adopt protein acetylation/deacetylation in metabolic regulation. Deacetylation of p53 prompts its ubiquitination and subsequent degradation, thereby promoting survival, and class III deacetylases also regulate several antioxidant enzymes. Dox significantly suppressed several SIRT deacetylases in the myocardium, abolishing antioxidants and exacerbating ROS production and apoptosis [80][88]. Upregulating the SIRT enzymes restores p53 ubiquitination, reduces caspase-3 activation, promotes Nrf2 and antioxidant enzymes, attenuates Dox-induced oxidative stress, and salvages DIC [80][81][82][88,89,90]. Interestingly, endothelial cells also highly express the SIRT enzymes [83][91]. Overall, the SIRT enzymes regulate cardiomyocytes’ metabolism, redox, and genomic integrity, all of which are aspects impaired in Dox-induced cardiotoxicity. Despite these findings, there remains a lack of SIRT activators developed for clinical therapy.
7. Dox Impairs Autophagy
2-Hydroxypropyl-β-cyclodextrin (HPβCD), a proposed treatment for Niemann–Pick type C disease characterized by impaired lipid metabolism, is an activator of the transcription factor TFEB that upregulates autophagy [84][92]. Co-treatment of HPβCD in Dox-treated neurons, thereby activating autophagy, attenuates peroxisome-associated ROS accumulation, reducing neurotoxicity [85][93].
All cells continually engage in autophagy (“self-eating”), an essential process that encompasses the encapsulation of the cell’s own macromolecules and organelles in the cytoplasm, followed by the lysosomal digestion of these biowastes into metabolic materials (e.g., amino acids, nucleic acids, phospholipids, etc.) [86][87][94,95]. Biowastes of autophagy include mRNAs, turnover proteins, toxic misfolded aggregates, desensitized receptors, and dysfunctional organelles such as abnormally proliferated mitochondria and peroxisomes. Disruption of autophagy leads to accumulating cytotoxicity, resulting in dysfunction and rapidly incapacitating metabolically stressed cells. As apoptosis is an energy-demanding process, this resulting cytotoxicity likely prompts necrosis and tissue inflammation, culminating in organ failures [88][89][96,97].
Autophagy is an emerging field in DIC research. Dox impairs autophagic flux at all stages. Dox inhibits various stress response factors that inhibit mTOR [90][98], thereby inhibiting autophagy. Dox-treated myocardium shows reduced numbers of autophagosomes and autolysosomes. On the other hand, Dox can also inhibit mTOR itself or modulate intermediate mediators, leading to excessive autophagosomes and autolysosomes, thereby dysregulating autophagy [79][91][87,99]. Dox-induced ROS accumulation also impairs lysosomal acidification and enzyme activity, resulting in the accumulation of autolysosomes [79][87]. Dysregulated autophagy, in turn, leads to mitochondrial dysfunction, impaired cellular metabolism, and apoptosis.
8. Dox Induces Sarcoplasm Leakage
Under Dox exposure, the myocardium exhibits cellular swelling, cytoplasmic vacuolation, myofibril disruption, and other characteristics of cardiac dysfunction [92][100]. Most notable is the severe dilation of the sarcoplasm (SR), followed by Ca2+ leakage into the cytoplasm, upsetting the cell ion-tonicity, impairing contractility, and potentiating ROS production [93][101]. In cardiomyocytes, Dox can increase CaMKII phosphorylation, which promotes the opening of RyR2 clusters on the SR, enabling Ca2+ leakage [94][102].
9. Dox-Induced Endotheliotoxicity (DIE)
The heart is a complex multicellular organ, comprising of cardiomyocytes—the main parenchymal cells behind cardiac pace-making and atrial/ventricular blood pumping—ECs (5% lymphatic, 95% vascular); vascular smooth muscle cells; fibroblasts; and pericytes. Cardiac ECs lining the endocardium and coronary vessels are known to establish regulatory cross-talk with other cell type populations to regulate vasomotor tone, blood flow, and angiogenesis [95][103], thereby being subjected to a high-energy oxidative metabolic environment, mitochondrial and peroxisomal redox regulation, and lipid oxidation, which influence cardiac function [96][97][104,105].
Virtually all cytotoxic aspects through which cardiomyocytes are subjected to Dox’s cytotoxicity also apply to endothelial cells (ECs). Since the ratio of ECs to cardiomyocytes in cardiac tissues is ~1.5:1, ECs are about 1.5-fold more susceptible to Dox’s genomic and oxidative impacts than cardiomyocytes [98][106]. Therefore, a comprehensive understanding of the mechanisms that alter EC function, leading to DIC, is warranted.
Recent findings have linked Dox-induced endothelial dysfunction, or Dox-induced endotheliotoxicity (DIE), to the susceptibility and severity of DIC. In this sense, both DIE and DIC may in fact contribute cumulatively to cardiovascular failure (Figure 2). Moreover, ECs are the first surface to interact with all circulatory entities, thereby the first to interact with Dox following systemic administration [99][107].
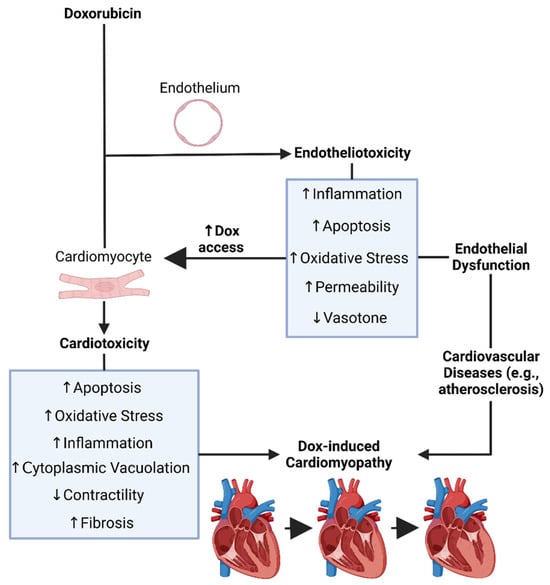
Figure 2. Doxorubicin’s induced endotheliotoxicity exacerbates cardiotoxicity. It was created with biorender.com (accessed on 26 December 2023). Dox = Doxorubicin.