Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Negrut Nicoleta.
COVID-19 was responsible for the latest pandemic, shaking and reshaping healthcare systems worldwide. Its late clinical manifestations make it linger in medical memory as a debilitating illness over extended periods. Long COVID is a complicated and multidimensional illness that affects a large proportion of those recovering from an acute COVID-19 infection. It has been linked to a variety of symptoms and problems, including chronic fatigue, cognitive impairment, respiratory troubles, cardiovascular irregularities, and psychological discomfort.
- Long COVID
- COVID-19
- SARS-CoV-2
- hypercoagulability
- dysbiosis
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causes the disease we now call COVID-19. With a decrease in deaths and hospitalizations due to acute COVID-19, interest has now slowly shifted towards the long-term complications seen in both prolonged and recovered cases. Thus, the term Long COVID has emerged, encompassing multiple symptoms that manifest after a SARS-CoV-2 infection. Long COVID places additional pressure on organs that are already strained by the acute form of the disease, resulting in multi-systemic long-term symptoms. Long COVID, according to the World Health Organization (WHO), represents any clinical manifestation present in the first three months after acute COVID-19, minimally evolving over two months, without another identifiable cause [1].
The symptomatology comprises a wide variety of symptoms, some more frequently encountered, such as fatigue, exertional dyspnea, insomnia, malaise, cognitive impairment, myalgia, cough, anosmia, arthralgia, chest pain, fever, tachycardia, and palpitations [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16][2][3][4][5][6][7][8][9][10][11][12][13][14][15][16]. Accurately establishing the epidemiological risk factors is highly important, as they are extremely valuable both for diagnostic and research purposes. Most of the articles that were reviewed link a high incidence of manifestation or persistence of Long COVID symptoms in females [2,3,4,5,6,7,8[2][3][4][5][6][7][8][9][10][11][12][14][15],9,10,11,12,14,15], older patients [4[4][7][15],7,15], patients with previous pulmonary diseases [4[4][7][9][10][15],7,9,10,15], diabetic patients [15[15][16],16], obese patients [3[3][4][15],4,15], patients with previous cardiovascular pathology [15[15][16],16], patients with increased SARS-CoV-2 viral load [8], smokers [3[3][4],4], patients with depression [4,5,6[4][5][6][7][8][9][10][11],7,8,9,10,11], and patients that experienced severe COVID-19 [7,8,9,13,14][7][8][9][13][14].
2. Pulmonary Involvement in Long COVID
The main impact of SARS-CoV-2 infection is on the respiratory system; the entrance of the virus into the epithelial lining of the lungs is facilitated by pneumonocytes that express the angiotensin-converting enzyme 2 (ACE2) receptors [17]. Upon infection by SARS-CoV-2, lung epithelial cells, which serve as the primary origin of inflammatory cytokines, engage in interactions with immune cells that have been recruited to the site of infection. This interaction plays a significant part in the development of inflammatory lung damage and subsequent respiratory failure [18]. The persistence and unregulated activation of cytokine storms can result in the impairment of the epithelial barrier. In the presence of such circumstances, the progression of lung fibrosis occurs due to the impairment of lung epithelial regeneration [19].
The injury inflicted by SARS-CoV-2 on a range of lung epithelial cells is attributed to the ACE2 receptors or transmembrane serine protease 2 (TMPRSS2) expression. ACE2 has been observed to be expressed in various types of cells in the airway epithelium, such as basal cells, ciliated cells, mucous cells, club cells, and intermediate cells [20]. According to a single-cell ribonucleic acid (RNA) sequencing analysis, it was observed that the average amount of ACE2 was comparatively higher in mucous cells as compared to other types of epithelial cells [21]. Alveolar type 1 (AT1) and alveolar type 2 (AT2) cells are susceptible to SARS-CoV-2 infection, with AT2 cells being the initial target [22]. Nonetheless, academic studies propose that a minor fraction of AT2 cells exhibit the expression of ACE2, while the expression of TMPRSS2 is minimal in the basal cells of the undifferentiated airway epithelium and more prominently expressed in the differentiated airway epithelium [23,24,25][23][24][25]. This proposed mechanism is accountable for the susceptibility of the lungs to COVID-19.
Dyspnea is the most common pulmonary symptom attributed to Long COVID. Dyspnea is defined as an unpleasant and unmanageable pattern of breathing related to an inability to ventilate well enough to provide the required amount of air for one normal breath. According to Hentsch L. et al., in their research published in 2021, three possible mechanisms were proposed for dyspnea induced by COVID-19 [26]. One of the mechanisms implies the interruption of afferent sensory signaling pathways by SARS-CoV-2, which may result in the inability of cortical structures responsible for processing the sensory aspects of dyspnea to receive afferent inputs from the brainstem. It is plausible that the virus may cause direct harm to the mechano- or irritant receptors located in the respiratory tract and/or chest wall, thereby impeding the transmission of afferent signals to the brainstem and higher brain structures. The second mechanism pertains to the possibility that SARS-CoV-2 may impede the capacity of cortical structures to identify or manage incoming sensory signals related to breathlessness originating from the brainstem. The occurrence of the conditions may arise either because of the direct impact of SARS-CoV-2 on nervous tissue or indirectly through the manifestation of inflammatory acute encephalopathy or cerebrovascular complications, including but not limited to ischemic or hemorrhagic stroke. The last mechanism implies that the cortical structures involved in the perception of dyspnea may exhibit a facilitative influence on the perception of breathlessness, like that of pain, which may be disrupted by SARS-CoV-2. The induction of dyspnea-related panic attacks through experimental inhalation of 35% CO2 has been demonstrated in patients with bilateral amygdala damage [27]. The self-reported levels of panic and fear in this group were found to be remarkably higher than those acknowledged by the comparison group with intact neurological function. The findings of this study may indicate that the activation of extra-limbic brain structures by CO2 occurs directly, and subsequently, these structures are controlled in a downward manner by the amygdala [27].
Several mechanisms of pulmonary damage in COVID-19 have been identified, with viral and immune-mediated pathways being implicated. Pulmonary fibrosis may arise because of chronic inflammation or as an idiopathic, age-related fibroproliferative process that is influenced by genetic factors [28]. Pulmonary fibrosis is a recognized consequence of acute respiratory distress syndrome (ARDS). Nevertheless, the clinical relevance of persistent radiological abnormalities after ARDS is limited and has decreased with the implementation of protective lung ventilation techniques [29]. Research has indicated that a significant proportion of individuals diagnosed with COVID-19, specifically 40%, are prone to developing ARDS. Furthermore, it has been observed that 20% of ARDS cases are classified as severe [30]. The manifestation of post-COVID-19 fibrosis will require further observation; however, initial examination of patients with COVID-19 upon hospital release indicates that over 33% of cured individuals exhibit fibrotic irregularities.
The defining characteristic of ARDS is the presence of diffuse alveolar damage (DAD). This is marked by an initial phase of acute inflammatory exudation, which is characterized by the presence of hyaline membranes. This is then followed by an organizing phase and a fibrotic phase [31]. Prior research has emphasized the significance of the duration of illness as a crucial factor in the development of pulmonary fibrosis after ARDS. The findings of this investigation indicate that a small proportion of patients (4%) who had a disease duration of less than one week, a notably larger proportion (24%) of patients with an illness lasting between 1 and 3 weeks, and a majority (61%) of patients who had a disease duration exceeding three weeks experienced the development of fibrosis [32]. The development and progression of pulmonary fibrosis may be instigated and facilitated by a cytokine storm resulting from an atypical immune response. The release of matrix metalloproteinases during the inflammatory phase of ARDS leads to epithelial and endothelial injury. The process of fibrosis involves the participation of vascular endothelial growth factor and cytokines, including interleukin (IL) 6 and tumor necrosis factor (TNF) a.
The etiology behind the differential outcomes of individuals who either recovered from an insult or developed progressive pulmonary fibrosis characterized by the accumulation of fibroblasts and myofibroblasts, along with excessive collagen deposition, remains unclear [33]. While COVID-19-induced ARDS appears to be the primary indicator of pulmonary fibrosis, various studies have indicated that it differs from classical ARDS in terms of its high and low elastance types.
The computed tomography (CT) results of numerous cases of COVID-19 do not indicate classical ARDS. In addition, the presence of abnormal coagulopathy is a notable pathological characteristic of this disorder. The mechanism underlying pulmonary fibrosis in COVID-19 differs from that observed in other fibrotic lung diseases, such as idiopathic pulmonary fibrosis (IPF). Notably, pathological observations suggest that the site of injury in COVID-19-induced pulmonary fibrosis is primarily the alveolar epithelial cells rather than the endothelial cells.
The act of coughing is a reflexive action that requires minimal conscious control. This reflex is initiated by the activation of peripheral sensory nerves that transmit signals to the vagus nerves. These nerves, in turn, provide sensory input to the brainstem at the solitary nucleus and the spinal trigeminal nucleus. The phenomenon of cough hypersensitivity has been established in the context of chronic cough, wherein the pathways responsible for coughing are believed to have undergone sensitization due to an increase in the magnitude of afferent signals transmitted to the brainstem. Coronaviruses, including SARS-CoV-2, gain access to host cells through specific receptors and proteases, namely ACE2, TMPRSS2, and furin [34]. SARS-CoV-2 may have the ability to directly interact with sensory neurons, as evidenced by the prevalence of sensory dysfunction, such as coughing, as well as olfactory and taste impairments, among individuals who have been infected with the virus [35]. The expression of ACE2 or TMPRSS2 in human airway vagal sensory neurons and their susceptibility to SARS-CoV-2 infection remain unknown. The bronchopulmonary vagal sensory neurons in mice were subjected to single-cell sequencing, which revealed the absence of murine ACE2 expression [36].
The potential involvement of supplementary viral entry factors in the interplay between SARS-CoV-2 and neurons cannot be disregarded. One such factor is neuropilin-1, which is present in vagal and other sensory neurons [37]. The study by D. H. Brann et al. conducted a sequencing analysis of human olfactory mucosal cells, revealing the absence of ACE2 and TMPRSS2 in olfactory epithelial neurons [38]. However, a significant expression of these genes was observed in support cells of the olfactory epithelium and stem cells [38]. The veracity of the results was validated through the cellular histological localization of ACE2 in the specialized neuroepithelium of supporting cells surrounding neuronal dendritic projections. It is noteworthy that the neuroepithelium comprises odor-sensing cilia [39]. Hence, it is plausible that the onset of anosmia resulting from SARS-CoV-2 infection could be attributed to the impact of the infected epithelium on neuronal function.
The ACE2 gene has been identified in a specific group of sensory neurons located in the thoracic ganglia of humans. These neurons are also known to provide innervation to the lungs. It is worth noting that a particular group of nociceptive neurons, which express calcitonin-related polypeptide alpha (CALCA) or purinergic receptor P2X 3 (P2RX3), have been found to exhibit expression [40]. These neuronal subtypes are like those found in the vagal sensory ganglia, which play an important role in triggering coughing. The similarity in developmental lineage and molecular phenotype between certain vagal sensory neurons, particularly those implicated in cough and dorsal root ganglion neurons, suggests a potential correlation between ACE2 expression in human vagal sensory neurons.
The etiology of persistent cough following SARS-CoV-2 infection is presently unknown, despite the possibility that the involvement of dorsal root ganglion neurons that contain nociceptors could account for the joint and chest pain, headache, and dyspnea symptoms experienced with Long COVID. The S1 spike protein of SARS-CoV-2 can cross the blood–brain barrier (BBB) in mice through absorptive transcytosis, indicating that a functional virus is not necessary for brain involvement [41]. Additional research is required to explore the potential direct interactions between the virus and the nervous system in the development of cough and other sensory symptoms in individuals with SARS-CoV-2 infection.
The exclusion of pathological or structural causes is crucial in the clinical management of chronic cough following COVID-19. This includes assessing fibrosis damage to lung parenchyma or damage to the airways resulting from SARS-CoV-2 infection or critical care management [42]. The presence of lung parenchymal alterations is a frequent observation on computed tomography (CT) scans in adult individuals affected by COVID-19. Additionally, a proportion of 10–20% of patients may experience the development of lung fibrotic changes [42]. The presence of lung fibrosis has been found to potentially heighten the sensitivity of the cough reflex in reaction to mechanical stimuli applied to the chest wall, as evidenced in individuals diagnosed with idiopathic pulmonary fibrosis [43].
The presence of a persistent cough in individuals experiencing post-COVID symptoms may be attributed to neuroinflammation, resulting in a state of heightened laryngeal and cough hypersensitivity. This phenomenon serves as the underlying cause of chronic refractory or unexplained cough [44,45][44][45]. Neuromodulators such as gabapentin and pregabalin have demonstrated efficacy in managing chronic refractory cough. The aforementioned strategy could be deemed as a viable option for addressing Long COVID, as these pharmaceutical agents may have utility in mitigating additional symptoms that coincide with coughing, such as discomfort, albeit with the possibility of exacerbating any cognitive impairment. The pulmonary manifestations of Long COVID are summarized in Figure 1.
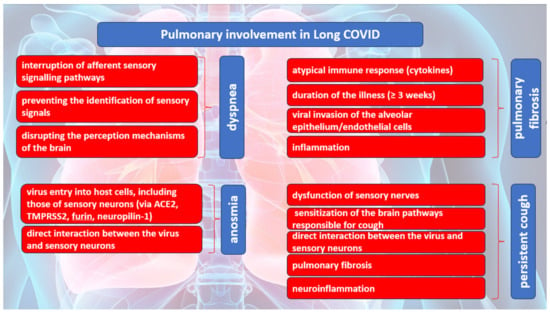
Figure 1. The causes of the most common pulmonary manifestations in Long COVID.
References
- Soriano, J.B.; Murthy, S.; Marshall, J.C.; Relan, P.; Diaz, J.V. A clinical case definition of post-COVID-19 condition by a Delphi consensus. Lancet Infect. Dis. 2022, 22, e102–e107.
- Sigfrid, L.; Drake, T.M.; Pauley, E.; Jesudason, E.C.; Olliaro, P.; Lim, W.S.; Gillesen, A.; Berry, C.; Lowe, D.J.; McPeake, J.; et al. Long COVID in Adults Discharged from UK Hospitals after COVID-19: A Prospective, Multicentre Cohort Study Using the ISARIC WHO Clinical Characterisation Protocol. Lancet Reg. Health-Eur. 2021, 8, 100186.
- Desgranges, F.; Tadini, E.; Munting, A.; Regina, J.; Filippidis, P.; Viala, B.; Karachalias, E.; Suttels, V.; Haefliger, D.; Kampouri, E.; et al. Post-COVID-19 Syndrome in Outpatients: A Cohort Study. J. Gen. Intern. Med. 2022, 37, 1943–1952.
- Subramanian, A.; Nirantharakumar, K.; Hughes, S.; Myles, P.; Williams, T.; Gokhale, K.M.; Taverner, T.; Chandan, J.S.; Brown, K.; Simms-Williams, N.; et al. Symptoms and Risk Factors for Long COVID in Non-Hospitalized Adults. Nat. Med. 2022, 28, 1706–1714.
- Pelà, G.; Goldoni, M.; Solinas, E.; Cavalli, C.; Tagliaferri, S.; Ranzieri, S.; Frizzelli, A.; Marchi, L.; Mori, P.A.; Majori, M.; et al. Sex-Related Differences in Long-COVID-19 Syndrome. J. Women’s Health 2022, 31, 620–630.
- Gebhard, C.E.; Sütsch, C.; Bengs, S.; Todorov, A.; Deforth, M.; Buehler, K.P.; Meisel, A.; Schuepbach, R.A.; Zinkernagel, A.S.; Brugger, S.D.; et al. Understanding the Impact of Sociocultural Gender on Post-Acute Sequelae of COVID-19: A Bayesian Approach. MedRxiv 2021.
- Tleyjeh, I.M.; Saddik, B.; Ramakrishnan, R.K.; AlSwaidan, N.; AlAnazi, A.; Alhazmi, D.; Aloufi, A.; AlSumait, F.; Berbari, E.F.; Halwani, R. Long Term Predictors of Breathlessness, Exercise Intolerance, Chronic Fatigue and Well-Being in Hospitalized Patients with COVID-19: A Cohort Study with 4 Months Median Follow-Up. J. Infect. Public Health 2022, 15, 21–28.
- García-Abellán, J.; Padilla, S.; Fernández-González, M.; García, J.A.; Agulló, V.; Andreo, M.; Ruiz, S.; Galiana, A.; Gutiérrez, F.; Masiá, M. Antibody Response to SARS-CoV-2 Is Associated with Long-Term Clinical Outcome in Patients with COVID-19: A Longitudinal Study. J. Clin. Immunol. 2021, 41, 1490–1501.
- Asadi-Pooya, A.A.; Akbari, A.; Emami, A.; Lotfi, M.; Rostamihosseinkhani, M.; Nemati, H.; Barzegar, Z.; Kabiri, M.; Zeraatpisheh, Z.; Farjoud-Kouhanjani, M.; et al. Risk Factors Associated with Long COVID Syndrome: A Retrospective Study. Iran. J. Med. Sci. 2021, 46, 428–436.
- Munblit, D.; Bobkova, P.; Spiridonova, E.; Shikhaleva, A.; Gamirova, A.; Blyuss, O.; Nekliudov, N.; Bugaeva, P.; Andreeva, M.; DunnGalvin, A.; et al. Incidence and Risk Factors for Persistent Symptoms in Adults Previously Hospitalized for COVID-19. Clin. Exp. Allergy 2021, 51, 1107–1120.
- Fernández-de-las-Peñas, C.; Martín-Guerrero, J.D.; Pellicer-Valero, Ó.J.; Navarro-Pardo, E.; Gómez-Mayordomo, V.; Cuadrado, M.L.; Arias-Navalón, J.A.; Cigarán-Méndez, M.; Hernández-Barrera, V.; Arendt-Nielsen, L. Female Sex Is a Risk Factor Associated with Long-Term Post-COVID Related-Symptoms but Not with COVID-19 Symptoms: The LONG-COVID-EXP-CM Multicenter Study. J. Clin. Med. 2022, 11, 413.
- Bai, F.; Tomasoni, D.; Falcinella, C.; Barbanotti, D.; Castoldi, R.; Mulè, G.; Augello, M.; Mondatore, D.; Allegrini, M.; Cona, A.; et al. Female Gender Is Associated with “Long COVID” Syndrome: A Prospective Cohort Study. Clin. Microbiol. Infect. 2021, 28, 611.e9–611.e16.
- Chudzik, M.; Lewek, J.; Kapusta, J.; Banach, M.; Jankowski, P.; Bielecka-Dabrowa, A. Predictors of Long COVID in Patients without Comorbidities: Data from the Polish Long-COVID Cardiovascular (PoLoCOV-CVD) Study. J. Clin. Med. 2022, 11, 4980.
- Chudzik, M.; Babicki, M.; Kapusta, J.; Kałuzińska-Kołat, Ż.; Kołat, D.; Jankowski, P.; Mastalerz-Migas, A. Long-COVID Clinical Features and Risk Factors: A Retrospective Analysis of Patients from the STOP-COVID Registry of the PoLoCOV Study. Viruses 2022, 14, 1755.
- Notarte, K.I.; de Oliveira, M.H.S.; Peligro, P.J.; Velasco, J.V.; Macaranas, I.; Ver, A.T.; Pangilinan, F.C.; Pastrana, A.; Goldrich, N.; Kavteladze, D.; et al. Age, Sex and Previous Comorbidities as Risk Factors Not Associated with SARS-CoV-2 Infection for Long COVID-19: A Systematic Review and Meta-Analysis. J. Clin. Med. 2022, 11, 7314.
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical Course and Risk Factors for Mortality of Adult Inpatients with COVID-19 in Wuhan, China: A Retrospective Cohort Study. Lancet 2020, 395, 1054–1062.
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-Converting Enzyme 2 (ACE2) as a SARS-CoV-2 Receptor: Molecular Mechanisms and Potential Therapeutic Target. Intensive Care Med. 2020, 46, 586–590.
- Thorne, L.G.; Reuschl, A.; Zuliani-Alvarez, L.; Whelan, M.V.X.; Turner, J.; Noursadeghi, M.; Jolly, C.; Towers, G.J. SARS-CoV-2 Sensing by RIG-I and MDA5 Links Epithelial Infection to Macrophage Inflammation. EMBO J. 2021, 40, e107826.
- Zhao, F.; Ma, Q.; Yue, Q.; Chen, H. SARS-CoV-2 Infection and Lung Regeneration. Clin. Microbiol. Rev. 2022, 35, e0018821.
- Zhang, H.; Rostami, M.R.; Leopold, P.L.; Mezey, J.G.; O’Beirne, S.L.; Strulovici-Barel, Y.; Crystal, R.G. Expression of the SARS-CoV-2 ACE2 Receptor in the Human Airway Epithelium. Am. J. Respir. Crit. Care Med. 2020, 202, 219–229.
- Duclos, G.E.; Teixeira, V.H.; Autissier, P.; Gesthalter, Y.B.; Reinders-Luinge, M.A.; Terrano, R.; Dumas, Y.M.; Liu, G.; Mazzilli, S.A.; Brandsma, C.-A.; et al. Characterizing Smoking-Induced Transcriptional Heterogeneity in the Human Bronchial Epithelium at Single-Cell Resolution. Sci. Adv. 2019, 5, eaaw3413.
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H.; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446.e14.
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-Cell RNA-Seq Data Analysis on the Receptor ACE2 Expression Reveals the Potential Risk of Different Human Organs Vulnerable to 2019-NCoV Infection. Front. Med. 2020, 14, 185–192.
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035.e19.
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 Receptor ACE 2 and TMPRSS 2 Are Primarily Expressed in Bronchial Transient Secretory Cells. EMBO J. 2020, 39, e105114.
- Hentsch, L.; Cocetta, S.; Allali, G.; Santana, I.; Eason, R.; Adam, E.; Janssens, J.-P. Breathlessness and COVID-19: A Call for Research. Respiration 2021, 100, 1016–1026.
- Coccia, C.B.I.; Palkowski, G.H.; Schweitzer, B.; Motsohi, T.; Ntusi, N. Dyspnoea: Pathophysiology and a Clinical Approach. S. Afr. Med. J. 2016, 106, 32.
- Liu, J.; Zheng, X.; Tong, Q.; Li, W.; Wang, B.; Sutter, K.; Trilling, M.; Lu, M.; Dittmer, U.; Yang, D. Overlapping and Discrete Aspects of the Pathology and Pathogenesis of the Emerging Human Pathogenic Coronaviruses SARS-CoV, MERS-CoV, and 2019-NCoV. J. Med. Virol. 2020, 92, 491–494.
- Burnham, E.L.; Janssen, W.J.; Riches, D.W.H.; Moss, M.; Downey, G.P. The Fibroproliferative Response in Acute Respiratory Distress Syndrome: Mechanisms and Clinical Significance. Eur. Respir. J. 2013, 43, 276–285.
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients with Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 1031.
- Liu, X.; Zhou, H.; Zhou, Y.; Wu, X.; Zhao, Y.; Lu, Y.; Tan, W.; Yuan, M.; Ding, X.; Zou, J.; et al. Risk Factors Associated with Disease Severity and Length of Hospital Stay in COVID-19 Patients. J. Infect. 2020, 81, e95–e97.
- Rai, D.K.; Sharma, P.; Kumar, R. Post COVID 19 Pulmonary Fibrosis—Is It Reversible? Indian J. Tuberc. 2020, 68, 330–333.
- George, P.M.; Wells, A.U.; Jenkins, R.G. Pulmonary Fibrosis and COVID-19: The Potential Role for Antifibrotic Therapy. Lancet Respir. Med. 2020, 8, 807–815.
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell Entry Mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734.
- Menni, C.; Valdes, A.M.; Freidin, M.B.; Sudre, C.H.; Nguyen, L.H.; Drew, D.A.; Ganesh, S.; Varsavsky, T.; Cardoso, M.J.; El-Sayed Moustafa, J.S.; et al. Real-Time Tracking of Self-Reported Symptoms to Predict Potential COVID-19. Nat. Med. 2020, 26, 1037–1040.
- Mazzone, S.B.; Tian, L.; Moe, A.A.K.; Trewella, M.W.; Ritchie, M.E.; McGovern, A.E. Transcriptional Profiling of Individual Airway Projecting Vagal Sensory Neurons. Mol. Neurobiol. 2020, 57, 949–963.
- Davies, J.; Randeva, H.; Chatha, K.; Hall, M.; Spandidos, D.; Karteris, E.; Kyrou, I. Neuropilin-1 as a New Potential SARS-CoV-2 Infection Mediator Implicated in the Neurologic Features and Central Nervous System Involvement of COVID-19. Mol. Med. Rep. 2020, 22, 4221–4226.
- Brann, D.H.; Tsukahara, T.; Weinreb, C.; Lipovsek, M.; den Berge, K.V.; Gong, B.; Chance, R.; Macaulay, I.C.; Chou, H.-J.; Fletcher, R.B.; et al. Non-Neuronal Expression of SARS-CoV-2 Entry Genes in the Olfactory System Suggests Mechanisms Underlying COVID-19-Associated Anosmia. Sci. Adv. 2020, 6, eabc5801.
- Chen, M.; Shen, W.; Rowan, N.R.; Kulaga, H.; Hillel, A.; Ramanathan, M.; Lane, A.P. Elevated ACE-2 Expression in the Olfactory Neuroepithelium: Implications for Anosmia and Upper Respiratory SARS-CoV-2 Entry and Replication. Eur. Respir. J. 2020, 56, 2001948.
- Shiers, S.; Ray, P.R.; Wangzhou, A.; Sankaranarayanan, I.; Tatsui, C.E.; Rhines, L.D.; Li, Y.; Uhelski, M.L.; Dougherty, P.M.; Price, T.J. ACE2 and SCARF Expression in Human Dorsal Root Ganglion Nociceptors: Implications for SARS-CoV-2 Virus Neurological Effects. Pain 2020, 161, 2494–2501.
- Rhea, E.M.; Logsdon, A.F.; Hansen, K.M.; Williams, L.M.; Reed, M.J.; Baumann, K.K.; Holden, S.J.; Raber, J.; Banks, W.A.; Erickson, M.A. The S1 Protein of SARS-CoV-2 Crosses the Blood–Brain Barrier in Mice. Nat. Neurosci. 2020, 24, 368–378.
- Ojha, V.; Mani, A.; Pandey, N.N.; Sharma, S.; Kumar, S. CT in Coronavirus Disease 2019 (COVID-19): A Systematic Review of Chest CT Findings in 4410 Adult Patients. Eur. Radiol. 2020, 30, 6129–6138.
- Jones, R.M.; Hilldrup, S.; Hope-Gill, B.D.; Eccles, R.; Harrison, N.K. Mechanical Induction of Cough in Idiopathic Pulmonary Fibrosis. Cough 2011, 7, 2.
- Ryan, N.M.; Birring, S.S.; Gibson, P.G. Gabapentin for Refractory Chronic Cough: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet 2012, 380, 1583–1589.
- Vertigan, A.E.; Kapela, S.L.; Ryan, N.M.; Birring, S.S.; McElduff, P.; Gibson, P.G. Pregabalin and Speech Pathology Combination Therapy for Refractory Chronic Cough. Chest 2016, 149, 639–648.
More