Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Péter Gál and Version 2 by Catherine Yang.
The complement system is the other major proteolytic cascade in the blood of vertebrates besides the coagulation-fibrinolytic system. Among the three main activation routes of complement, the lectin pathway (LP) has been discovered the latest, and it is still the subject of intense research. Uncontrolled complement activation can contribute to the progression of many diseases (e.g. stroke, kidney diseases, thrombotic complications, and COVID-19). In most cases the lectin pathway has also been implicated.
- complement lectin pathway
- pattern recognition
- protease
- complement-related diseases
1. Introduction and Brief History of the Lectin Pathway
The complement system is an important effector arm of the innate immune system. It consists of about 40 protein components, both fluid-phase and cell-surface molecules. This remarkable molecular network carries the properties typical of the immune system in general: it can recognize, label, and eliminate pathogens and the dangerously altered host cells. The recognition function is mediated by pattern recognition molecules (PRMs) such as C1q, MBL, and ficolins. These PRMs typically contain globular domains responsible for binding to dangerous structures and collagen-like arms to which serine protease components bind. From an enzymatic point of view, the system can be considered a proteolytic cascade system, like the blood coagulation and the fibrinolytic systems. These proteolytic cascade systems in the blood are evolutionary and functionally closely related, and they form a single proteolytic network [1]. For practical and didactical reasons, this unified network can be divided into separate cascade systems, but understandably, there is a lot of cross-talk between these systems.
Most of the serine proteases of the complement system are present as zymogens in the circulation. When the PRMs bind to the target surface, the associated serine proteases become activated and initiate the complement cascade by cleaving and activating the subsequent components. Since one active protease can cleave numerous zymogen molecules, the proteolytic cascade carries an enormous amplification potential. As a result, the complement activation is tightly regulated by various inhibitors to avoid self-tissue damage. Indeed, if the complement activation happens in an uncontrolled manner, it can contribute to the development and progression of serious disease conditions [2].
There are three ways through which the complement system can activate: the classical, the alternative and the lectin pathways. The classical pathway was discovered first, at the turn of the 19th and 20th centuries, and it determined the general view of the complement system for a long time. At that time, the system was considered an effector mechanism of the adaptive immune response, which “complements” the action of the antibodies. Later, in the 1950s, an antibody-independent activation pathway, the alternative pathway, was discovered, but it became generally accepted only in the 1970s. The alternative pathway clearly linked the complement system to innate immunity and demonstrated that this molecular system can independently recognize and destroy the pathogens. The lectin pathway is the latest discovered activation route of the complement system (Figure 1). It was already observed in the 1980s that the MBL, in a complex with proteases, can activate the lectin pathway [3][4][3,4]. Initially, it was thought that MBL, like C1q of the classical pathway, binds C1r and C1s and initiates the classical pathway activation. Later, it turned out that MBL is associated with another protease called MASP (MBL-associated serine protease, earlier known as P100), which is able to cleave C4 and C2 to generate the C3 convertase complex [5]. A few years later, however, it turned out that “MASP” is a mixture of two proteases (MASP-1 and MASP-2), and the protease present in much smaller amounts (MASP-2) is responsible for cleaving C4 and C2 [6]. It was also shown that isolated MASP-2 can autoactivate in vitro and initiate the lectin pathway activation without the contribution of any other protease [7]. The conclusion drawn from this observation, i.e., that MASP-2 alone is sufficient for activation, defined the theory of lectin pathway activation for more than a decade. In 2001, a third protease component, MASP-3 was discovered, which is a product of the MASP1 gene [8]. In the beginning, nothing was known about the biological role of MASP-3, and no physiological substrate was known either. For lack of a better theory, MASP-3 had been attributed an inhibitory role, despite the fact that it is an enzymatically active protease. Now, MASP-3 plays a major role in complement activation and beyond. In the meantime, other, non-enzymatic components of the lectin pathway were also discovered. From both MASP1 and MASP2 genes, shorter N-terminal fragments lacking the catalytic domain (MAp19 and MAp44) are expressed via alternative splicing or polyadenylation [9][10][9,10]. The number of PRMs also has been increased by the discovery of ficolins (ficolin-1, -2 and -3) [11] and other collectins (CL-K1, CL-L1) [12].
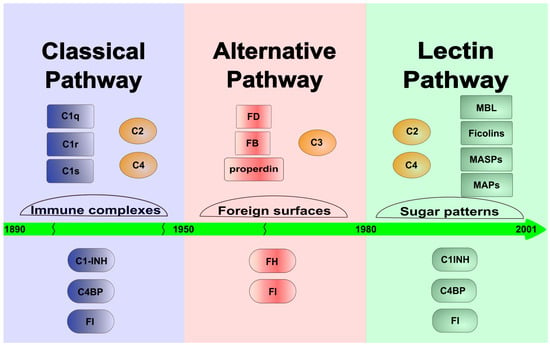
Figure 1. Timeline toward the discovery of the LP. The figure shows the time sequence of the discovery of the individual activation routes of the system. The classical pathway dominated the early 20th century, while the alternative pathway was described in the middle of that century. Finally, the lectin pathway was discovered and characterized at the end of the 20th and at the beginning of the 21st century. The figure shows the most important components (upper lane) and the most important fluid-phase inhibitors (lower lane) of each complement pathway.
The lectin pathway is perhaps the most complex activation pathway among the complement pathways; it has at least six PRMs, three serine proteases, and two non-catalytic fragments. Since the PRMs can have a different oligomerization status, from dimer to hexamer [13], and one PRM can bind different MASPs and MAps, many different activation complexes can form.
2. Diseases with Potential Lectin Pathway Involvement
Detrimental activation of the complement system is known to underlie many diseases. In most cases, more than one pathway is activated at the same time, and it is difficult to distinguish which plays a greater role in pathological processes. However, there are diseases in which the activation of the lectin pathway is of particular importance (Table 1).
Table 1.
Lectin pathway components involved in human diseases and LP-related drugs in clinical or preclinical trials.
| Disorders | LP Components Involved |
Drugs under Clinical Trial | Drug Candidates under Preclinical Trial |
|---|---|---|---|
| Renal disorders | |||
| IgA nephropathy | MBL, ficolin-2, MASP-1, MASP-2, MASP-3, MAP-19 [14][15][16][17][18][145,146,147,148,149] | Narsoplimab (recently discontinued) [19][20][150,151] |
Anti-MASP-3 (OMS906) [21][152] |
| Membranous nephropathy | MBL, MASP-1, MASP-2 [22][23][24][25][153,154,155,156] |
Narsoplimab [26][157] | |
| Diabetic kidney disease | MBL, ficolin-3 [27][28][158,159] | Anti-MBL MAb [29][160] | |
| Ischemia Reperfusion Injury | |||
| Renal IRI | MBL, CL-K1, MASP-2 [30][31][32[36][37][76,77][33][34],78[,16135,162],163,164,165] |
CL-K1 inhibition by L-fucose; C1INH; TFMI-2 [38][39][40][41][166,167,168,169] |
|
| Myocardial IRI | MBL, ficolin-2, MASP-1, MASP-2 [42][43][44][45][46[49][138,170][,17147][,17248,173],174,175,176] |
C1INH; anti-MBL MAb [50][51][177,178] | |
| Ischemic stroke | MBL, MASP-1, MASP-2 [52][53][54][55][179,180,181,182] | ||
| Atherosclerosis | MBL, ficolin-1, ficolin-2, ficolin-3, MASP-3 [56][57][58][183,184,185] | ||
| COVID-19 | MBL, ficolin-2, ficolin-3, MASP-2 [59][60][61][62][63][64][186,187,188,189,190,191] |
Narsoplimab * [65][192] | |
| Hereditary angioedema | MASP-1 [66][67][193,194] | C1INH (approved) ** [68][195] | |
| Schizophrenia | MBL, ficolin-2, MASP-2 [69][70][59,196] |
* Target: MASP-2; ** Main targets: kallikrein and FXIIa.
2.1. Renal Diseases
Probably the most affected part of human body by pathological complement activation is the kidney. Since kidney cells can produce complement components, the entire repertoire of the complement cascade is available locally, while blood circulation can also provide active complement elements. Unwanted activation of the lectin pathway is often not the primary driver of glomerular diseases but a consequence of the presence of injured kidney cells [71][197].
2.1.1. IgA Nephropathy
IgA nephropathy (IgAN) is a severe primary glomerulonephritis, which is a leading cause of renal failure. It is now treated as an autoimmune disease that is initiated by flawed IgA1 synthesis and abnormal glycosylation of the heavy chain. Galactose-deficient IgA1 accumulates in the circulation that leads to a complex formation with autoantibodies. Deposits develop in the glomeruli causing mesangial cell proliferation [72][198]. The most characteristic symptoms of IgAN are hematuria, proteinuria, and acute kidney injury. These processes eventually lead to chronic kidney diseases. The pivotal role of complement in the progression of the IgAN is well known, and glomerular co-deposition of IgA1 and complement components, mostly C3, have been observed in biopsy samples. Lectin pathway involvement has been proven via the detection of C4d deposition in renal biopsies without C1q present [14][145]. The initiation of the LP is triggered by pattern recognition molecules binding to the aberrantly glycosylated IgA1. Newly exposed N-acetylgalactoseamine serves as target to MBL and ficolin-2. Another explanation for LP activation can be the binding of MBL to neoepitopes on the surface of injured glomerular cells. MBL and MASP-1 have been detected in 24% of IgAN patients along with C3b/C3c and C5b9, but a correlation with the severity of IgAN could not be established [15][146]. Roos et al. also found MBL, ficolin-2, MASP-1, MASP-3 and C4d deposits in 25% of the patient group. These patients suffered more serious renal damage such as increased mesangial proliferation, glomerular sclerosis, interstitial infiltration, and proteinuria [16][147]. Data from a large Spanish cohort presented C4d deposits in 38.5% of the patients that correlated with the development of end-stage kidney disease [73][199]. Variations in LP components in the circulation were measured, but a correlation with IgAN severity was not consistent among different groups. Guo et al. [17][148] presented data that showed that low MBL levels indicated more serious IgAN, while Roos et al. [16][147] could not verify the connection. Plasma concentration of ficolins-1, -2, MBL, MASP-1 and MAP-19 significantly increased, while the level of MASP-3 decreased in IgAN patients. A low level of MASP-3 accompanied by an increased level of C3 cleavage products and FHR5 indicated progressive disease [18][149]. Clinical observations suggest that IgAN is mostly triggered by LP activation, while aggravation of the disease might be due to the participation of the AP.
2.1.2. Membranous Nephropathy
Aberrant glycosylation of IgG4 gives rise to the activation of the lectin pathway in another rare but serious glomerulonephritis, the membranous nephropathy (MN). The major activators in primary MN have been proven to be antibodies that bind to the phospholipase A2 receptor (PLAR2) on podocytes [26][157]. These IgG4 type antibodies do not activate the CP, but the co-localization of MBL, C3 and C4d on the subepithelial surface of glomerular capillaries indicates activation of the LP [22][153]. PLAR2-seropositive patients present elevated levels of MBL and C4d in serum and in urine, which seems to correlate with proteinuria [23][154]. It has been shown that patients with anti-PLAR2 antibodies are more prone to reach remission if they had initially lower serum levels of MBL, MASP-1 and MASP-2 [24][155]. Another MN-causing antigen is the thrombospondin type 1 domain-containing 7A (THSD7A). The LP-activating mechanism is similar to that of PLAR2. Human anti-THSD7A antibodies are able to raise the levels of MBL, MASP-1 and -2 in mice [25][156].
2.1.3. Diabetic Kidney Disease
One of the most common causes behind end-stage renal failure worldwide is the slow inflammation in the kidney caused by diabetes mellitus type 1 or 2. There is an enormous amount of data in the literature dealing with the role of the LP in diabetic kidney disease (DKD) [74][200]. Unwanted initiation of complement activation is due to the enzymatic or non-enzymatic glycosylation of proteins, lipids, and nucleic acids in the hyperglycemic environment. This phenomenon can affect the complement cascade in two ways. The first is the activation of the LP through MBL and ficolin-3 binding to these new neo-epitopes, while the other is the uncontrolled complement activity due to hyperglycosylated regulatory proteins losing their function. The data showed that levels of MBL, C4d, C3a and four other complement components significantly increased in the glomeruli, arterioles and urine of DKD patients compared to diabetic patients who did not suffer from renal damage [27][28][158,159].
2.2. Ischemia/Reperfusion Injury
2.2.1. Renal Ischemia/Reperfusion Injury
The vital role of complement is now generally accepted in ischemia reperfusion injury (IRI) in several organs such as the kidney, heart, intestine, liver, skeletal muscle, and brain. Ischemia develops when blood flow is permanently obstructed in a certain tissue that is followed by reperfusion when blood flow returns to the affected area. Ischemia and reperfusion initiate inflammation-like processes that cause severe tissue damage. Complement activation occurs at the early stage of IRI locally, and the activated complement components such as C3a, C5a and sC5b-9 appear in the circulation ahead of clinical symptoms [75][201]. The recruitment and activation of neutrophils by anaphylatoxins and the effect of MAC lead to the complement-mediated tissue injury that can spread to tissues initially not ischemic. Lectin pathway components, especially the pattern recognitions molecules collectin-11 (CL-K1), MBL and their enzymatic partner, MASP-2, seem to play key roles in commencing undesirable inflammation after renal and cardiac reperfusion [33][161]. In kidneys, LP activation is suggested to be triggered by CL-K1, which is expressed at elevated levels by tubule cells after IR stress. MASP-2 and C3d were found to be colocalized with CL-K1 and its preferred ligand L-fucose on the surface of renal epithelial cells [34][162]. An alternative activation of the LP is suggested by Yaseen et al. [32][78], the so-called C4-bypass where MASP-2 initiates the LP without the cleavage of the C4 protein [30][31][32][76,77,78]. The researchers found this theory suitable for the LP activation in kidneys during IRI based on an animal model. MBL deficiency proved to be protective in a renal mouse model [35][163], but C4 knockout mice were not protected from IRI [36][164]. Nevertheless, data are not available in human patients to further extend this hypothesis. Complement activation during kidney transplantation is an intensively examined area, since transplanted organs experience IRI during implantation. Clinical data have shown that recipients who possessed lower serum levels of MBL were prone to avoid organ rejection [37][165].
2.2.2. Myocardial Ischemia/Reperfusion Injury
Similar processes are initiated by the LP in the heart after myocardial infarction (MI). A shortage of blood supply generates damage-associated molecular patterns (DAMPs) and causes the damage of complement regulatory proteins on the endothelial cells, leading to the devastation of cardiomyocytes. LP activation plays an important part in the reperfusion phase of myocardial ischemia/reperfusion injury that is shown to increase the extent of infarct size by up to 50% [76][202]. The deposition of MBL and C3 in a rat heart was the first observation concerned with LP involvement [43][170], which was further demonstrated in other experimental models. LP activation undoubtedly plays a part in MI, but the limited number of patients makes human data less conclusive. According to Zhang et al., the level of MASP-2 in patients with open heart surgery dropped as low as 50% compared to those patients who had stable coronary artery disease (CAD) [44][171]. In another study, a lower concentration of MASP-2 and a higher concentration of MASP-1 were observed in myocardial infarction patients compared to the ones in CAD patients [45][172]. MASP-2 and ficolin-2 consumptions have been shown in several papers [46][47][173,174], but a correlation between the infarct size and the examined complement components could not be confirmed. The role of MBL is controversial. Bonnemeier et al. presented data that showed that 30-day mortality among MBL-deficient MI patients was significantly lower [48][175]. On the other hand, Holt et al. demonstrated that low levels of pattern recognition LP molecules provide no protection from the consequences of IRI. Furthermore, MBL and ficolin levels had no prognostic value concerning the short-term outcome of myocardial infarction [49][176]. MASP-1 might have an interesting part in the initiation of MI due to its clot generating ability [42][138]. Generally, more in vitro and in vivo data are needed to determine whether the activation of the LP is a cause or a consequence of myocardial infarction in humans.
2.2.3. Ischemic Stroke
Stroke is the third leading cause of death in developed countries. A sudden change in blood supply initiates hypoxia and ischemic injury in a part of the brain, leading to tissue damage and acute neurological disfunctions. Changes in the levels of complement components in patients after stroke indicate the initiation of the complement cascade [77][203]. MBL-null mice were used in focal cerebral IRI to examine the contribution of the LP to the severity of stroke. A smaller infarction size, better functional outcome, significantly decreased C3 deposition, and neutrophil infiltration were measured, while reconstitution with human recombinant MBL enhanced brain damage [52][179]. Furthermore, in the case of stroke patients, the outcome was associated with the MBL genotype. Low MBL levels also meant lower levels of C3, C4, CRP and proinflammatory cytokine profiles and better recovery after 3 months [52][179]. The significant role of MBL in ischemic stroke was further reinforced by data showing a smaller infarct size and a more favorable outcome in MBL-deficient patients who received conservative treatment [53][180]. LP involvement was shown in a MASP-2 knockout mouse model of focal cerebral ischemia. Mice lacking MASP-2 developed reduced neurological deficits and infarct volumes compared to wild-type mice [54][181]. Tsakanova et al. found a correlation between the increased activity of MASP-1/-2 and the risk of ischemic stroke [55][182]. In the patients’ blood, a more pronounced cleavage of C2 and C4 was detected compared to that detected in the healthy controls. They postulated that some genotypes had a protective role against the development of stroke and post-ischemic brain damage.
2.3. Atherosclerosis
More evidence supports the idea that lectin pathway activity is related to atherosclerosis and atherosclerosis-related acute myocardial infarction (AMI). Chronic low-level inflammation in the vessels contributes to the generation, progression and also the destabilization of plaques. The presence of MBL and ficolins in a plaque environment indicates LP involvement. Vengen et al. showed that MBL deficiency correlated with the severity of atherosclerosis [56][183]. More recently, ficolin-1, 2 and -3 were found in carotid plaques, while MBL was present mostly in ulcerated plaques, where MBL-driven LP activation was shown to be associated with plaque instability [57][184]. Furthermore, a higher concentration of ficolin-2 and pentraxin-3 and a low level of MASP-3 might predispose individuals to develop AMI [58][185].
2.4. COVID-19
The complement cascade usually plays a major role in diseases that cause extensive inflammation, as is what happens in COVID-19 [78][204]. Shortly after entering the host, structural proteins of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) appear in the circulation and in all body fluids, initiating an immense immune response. All three complement pathways proved to be active after the onset of COVID-19, and more severe symptoms correlated with more intense complement activation. Several findings have validated that the lectin pathway contributes in causing or aggravating symptoms, since activated LP components have appeared in plasma and biopsy samples of COVID patients. Increased plasma levels of the MASP-1/C1INH complex and C4d were shown in a large cohort, which indicated LP activation [59][186]. Furthermore, elevated MASP-2 concentration was measured by a specific ELISA in hospitalized patients that correlated with the levels of ficolin-2, -3 and the C-reactive protein [60][187]. The importance of MASP-2 was further demonstrated by its significant deposition in kidney and lung biopsy samples of deceased patients [61][188]. MBL is a central player in the interaction between the LP and viral proteins. Highly glycosylated spike (S) proteins can be recognized by MBL. Therefore, MBL-driven complement activity hinders virus entry at the early stage of infection [62][189]. On the other hand, MBL polymorphism does not influence the outcome of disease progression [59][186]. The most abundant viral protein in plasma is the nucleocapsid (N) protein which is thought to interfere with the LP. Gao et al. presented data that showed that the N-protein of SARS-CoV-2 could initiate the LP through binding directly to and activating zymogen MASP-2 [63][190]. They also showed that the N-protein was capable of enhancing the autoactivation of MASP-2. In vitro binding of MASP-2 and MBL to the N-protein was also demonstrated by Ali et al. [64][191]. Notwithstanding, these results are in contrast with the findings of Stravalaci et al. who could not confirm MBL and N-protein interaction [62][189]. According to the controversial experimental data and less conclusive clinical observations, the role of the LP in COVID-19 is still not fully understood.
2.5. Hereditary Angioedema (HAE)
Insufficient lectin pathway regulation by C1INH may contribute to the development of a rare but severe disease, hereditary angioedema (HAE). In HAE, mainly plasma kallikrein is responsible for the cleavage of high molecular weight kininogen and the release of the proinflammatory peptide, bradykinin. Kallikrein activity is regulated by C1INH, but mutations of the SERPING1 gene lead to low plasma C1INH activity that impairs the control of both the contact activation and the complement system. The accumulation of bradykinin results in increased vascular permeability and intense tissue swelling. The cleavage of high molecular weight kininogen by MASP-1 and the release of bradykinin were shown by Dobó et al. in human plasma [66][193]. This serves as an example of the connections between the kallikrein–kinin and the lectin cascades. Clinical data showed that levels of MASP-1 and MASP-1/C1INH complexes were reduced in HAE patients, which correlated with the severity of the disease. Significant consumptions of MASP-1 and C4d were considered to be caused by LP activation [67][194]. Another contributing factor of MASP-1 to HAE could be the increased permeability of endothelial cells due to the action of this protease on PARs, as discussed in the “Cellular effects of MASPs” section.
2.6. Autoimmune Diseases: Systemic Lupus Erythematosus and Rheumatoid Arthritis
Impaired control of the complement system may lead to autoimmune diseases [79][205]. Systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) are among them, and increasing data support the participation of the lectin pathway in the onset of their symptoms.
SLE is a chronic, life-threatening, and complex immune-mediated disorder. The deposition of activated complement products in the tissue and hypocomplementemia indicate massive complement involvement, mostly through the activation of the CP by autoantigens. The role of LP components has recently begun to be investigated, and the mechanism has not been fully revealed. The serum levels of LP proteins differ from those of healthy controls. Ficolin-2, ficolin-3, MASP-2, and MASP-3 concentrations were higher in a large cohort of SLE patients [80][206]. The level of MBL seemed to be significantly elevated and correlated with the severity of the disease Furthermore, the results indicated MBL participation in the development of lupus nephritis in SLE patients [81][207]. Another study showed reduced levels of MASP-1 and MASP-2 in proliferative lupus nephritis and severe SLE without lupus nephritis, while in patients with membranous nephritis, the concentration of these serine proteases was the same as in healthy controls [82][208]. Functional ELISA assays further reinforced the activation of the LP, measuring independently the activation of the three pathways in SLE serum samples [83][209]. Serum levels of MASP-1/C1INH and C1s/C1INH complexes indicated that both the CP and LP are active in SLE. However, LP contribution is more substantial in lupus nephritis [84][210].
Rheumatoid arthritis is characterized by chronic inflammation of the joints. Anomalous complement function due to faulty control contributes to an autoimmune disease that attacks cartilage, synovium and finally bones [85][211]. Synovial fluid that represents local inflammatory situations contains activated complement factors in elevated concentrations [86][212]. The accumulation of the IgG glycoform that lacks galactose initiates not only the CP, but the LP through MBL binding to newly exposed N-acetyl glucosamine in the Fc region [87][213]. Ammitzboll et al. measured a higher level of MBL, ficolin-1, ficolin-3, MASP-2 and MASP-3 in the plasma and synovial fluid of RA patients. Furthermore, ficolin-1 concentrations correlated with the disease activity [88][89][214,215]. A collagen antibody-induced mouse RA model demonstrated an interesting new role of liver-derived MASP-3, namely, its contribution to the damage of joints affected by RA [90][216].
2.7. Schizophrenia
Schizophrenia is a severe mental disorder, which causes a range of various psychological symptoms. Symptoms can include hallucinations, delusions and disorganized thinking. The etiology of schizophrenia is unknown, but it seems likely that a combination of genetic and environmental factors contributes to the development of the disease [91][217]. It has also been suggested that immunological processes [92][218], and particularly the inappropriate complement activation, may also play a role in the etiopathogenesis [93][219]. Recently, a number of observations have suggested that the complement system plays important roles in the brain’s development [94][220]. The components of the lectin pathway are expressed in the brain, and they are involved in the neuronal migration during brain development [95][221]. Malfunctioning of the lectin pathway may contribute to the development of schizophrenia. Mayilyan et al. showed that the functional activity of the MBL-MASP-2 complexes is higher in the sera of schizophrenic patients compared with healthy individuals [69][59]. Recently, the same authors also showed that a significant increase in ficolin-2-bound MASP-2 activity was observed in schizophrenia [70][196]. Although it is difficult to estimate the exact contribution of the disturbed lectin pathway to the pathogenesis of such a complex disease, these results clearly indicate that an intact complement system is needed for healthy neurological development and functions.
2.8. Influence of Aging
Since the lectin pathway fights against the invading pathogens before the development of the adaptive immune response, it is not surprising that an intact lectin pathway is more important in early childhood than in adulthood. Deficiency of the lectin pathway components can result in recurrent infections in children [96][97][98][222,223,224]. The age variations of the concentration of lectin pathway components are in agreement with the above phenomenon. In the case of MBL and ficolins, the highest serum concentrations were detected in children (typically 1–16 years), while in neonates and in adults, these concentrations were significantly lower [99][225]. Among adults, only insignificant changes of lectin pathway protein concentrations were observed with age. This indicates that aging does not influence lectin pathway activity in adulthood [100][39].