1. Reduced Cancer Risk in Genetic Syndromic Conditions
Patients with syndromic conditions have always been a population described in numerous studies relating to the co-occurrence of other morbidities. Among these studies, attempts were made to establish whether there was an increased risk of cancer in this patient population. Over time, this higher risk has been established for numerous syndromic conditions with a known genetic basis, such as overgrowth syndromes
[1][18], RASopathies
[2][19], phacomatosis
[3][20], and some microdeletions syndromes, among which is velocardiofacial syndrome
[4][21].
1.1. Down Syndrome
Down syndrome (DS) has been extensively studied in relation to the reduced incidence of solid tumors. From a superficial analysis, DS individuals have the same incidence of cancer as the rest of the normal population. However, a comprehensive study involving a large cohort of individuals with DS revealed a decreased risk of all major groups of malignant solid tumors, except for testicular cancer, while leukemia is more frequent, especially during the pediatric stage
[5][22]. Particularly noteworthy was the significantly lower occurrence of lung, skin, cervical, and female breast cancers
[6][23].
This phenomenon is remarkable, considering that individuals with DS possess several major risk factors that predispose them to cancer. These factors include hypotonia leading to reduced physical activity and obesity, accelerated organ aging, immunodeficiency, dysfunction in DNA repair systems resulting in increased DNA damage, mitotic chromosomal instability, mitochondrial alterations leading to high levels of reactive oxygen species (ROS), and elevated expressions of multiple oncogenes located on chromosome 21
[7][24].
Various hypotheses have been proposed to explain this lower risk of solid tumors, encompassing genetic dosage effects, the overexpression of tumor suppressor/repressor genes, disrupted metabolism, impaired neurogenesis and angiogenesis, increased apoptosis, dysregulation of the immune system, and epigenetic abnormalities.
The
ETS2 (V-Ets Avian Erythroblastosis Virus E26 Oncogene Homolog 2) gene, located on chromosome 21q22.3, regulates cell survival by controlling multiple targets such as TP53, P21, CYCLIND1, PRESENILIN1, and ICAM1
[8][25]. Initially categorized as a proto-oncogene linked to acute megakaryoblastic leukemia when involved in somatic balanced translocations
[7][24], a pivotal study utilizing mouse models of DS revealed that overexpression of
ETS2 in APC
min mice prevented the development of intestinal tumors
[8][9][10][25,26,27]. Despite it never being found mutated in tumor-predisposing conditions,
ETS2 appears to act more precisely as a tumor repressor rather than a suppressor gene. Notably, in the same study, different dosages of
ETS2 alleles in mice corresponded closely to the incidence of intestinal tumors in the APC
min background
[10][27].
Another gene,
SOD1, located in 21q22.11, contributes to disrupted metabolism by overexpressing superoxide dismutase 1, leading to heightened ROS levels in neurons, lymphocytes, and fibroblasts. The imbalance between the cytosolic isoform of
SOD1 (elevated) and glutathione peroxidase (normal) induces intracellular ROS accumulation, consequently causing oxidative stress, cell damage, and apoptosis
[11][12][28,29].
Some studies suggest that the overall impact on tumors may relate to disrupted angiogenesis, as individuals with DS often exhibit frequent benign or indolent neoplasms, but significantly fewer aggressive tumors
[13][30]. Among the potentially “protective genes” overexpressed in DS individuals interfering with angiogenesis, the prominent one is the
COL18A1 gene.
The
COL18A1 (Collagen Type XVIII α1 Chain) gene encodes the α chain of collagen XVIII, which, upon proteolysis, transforms into endostatin, a potent inhibitor of angiogenesis, by inhibiting the pro-angiogenic factor VEGFA (vascular endothelial growth factor A). Endostatin levels are increased in individuals with DS
[14][31]. Reduced angiogenesis could play a significant role in halting tumor progression, albeit of lesser importance in preventing its occurrence.
Special consideration must be given to the
DYRK1A (Dual-specificity tYrosine-phosphorylation Regulated Kinase 1A) gene, which is expressed ubiquitously and encodes a protein kinase involved in various aspects of the DS phenotype.
DYRK1A participates in the proliferation and differentiation of neuronal progenitor cells
[15][32]. It also plays a dual role in tumorigenesis, being overexpressed in numerous cancers.
DYRK1A promotes tumorigenesis
[16][33] by phosphorylating the NFAT transcription factors
[17][34], sustaining proliferation through upregulation of the RAS/MAPK signaling pathway, resisting apoptosis via caspase 9 phosphorylation, and enhancing angiogenesis through sustained accumulation of the VEGFR2 receptor
[18][35]. However, its pro-cancer ability is counteracted by tumor repressor properties mediated by phosphorylation of TP53 serine 15
[19][36], inducing cell cycle arrest in G0/G1, transcriptional suppression via the DREAM complex, and playing a central role in the double-strand break repair mechanism through RNF169 phosphorylation
[20][37].
The seemingly contradictory aspects of the DS phenotype are mirrored by the dual role of
DYRK1A. While mouse models exist that either overexpress
[21][38] or lack
Dyrk1a [22][39], most studies using these mice focus on the neurological aspects of DS. It would be intriguing to explore, in various cancer models, how
Dyrk1a dosage affects cancer development and progression.
In terms of the immune system, individuals with DS exhibit increased susceptibility to respiratory infections. Some propose that DS represents a primary immunodeficiency, due to global thymic hypofunction and lymphopenia of T and B cells. Paradoxically, despite this immune dysfunction predicting an increased cancer incidence, this does not occur. Some authors suggest this could be due to an increased proportion of γδT cells responsible for tumor immunosurveillance, favoring the early elimination of cancer cells
[23][40]. However, the gene or genes responsible for this phenotype remain unidentified.
Regarding epigenetic alterations, individuals with DS display an aberrant pattern of methylation across the entire genome, with areas of both hypermethylation and hypomethylation
[23][40]. Recurrent and reproducible epigenetic changes on chromosome 21 are observed in various tissues and cell subtypes.
Chromosome 21 contains up to 30 miRNAs, with five of them being overexpressed and linked to the DS phenotype: let-7c, miR-99a, miR-125b2, miR-155, and miR-802
[24][41]. Their overexpression leads to reduced expression of proteins encoded by the targeted mRNAs. If some of these proteins were oncogenic, this would protect individuals with DS from developing tumors. The overexpression of these miRNAs may, to some extent, explain the genome-wide impact of the extra chromosome 21.
Concerning the genetic dosage effect, the overexpression of certain genes has been examined. The S100B (S100 Calcium Binding Protein β) gene encodes a calcium-binding protein secreted by glial cells that induces neural cell differentiation. Its overexpression in DS promotes neurodegeneration
[25][42] but also exhibits a protective effect against neuroblastoma
[18][19][20][35,36,37], correlating with the absence of neuroblastoma in children with DS.
Additionally, individuals with DS manifest a premature aging phenotype across various tissues, experiencing increased genotoxic stress and oxidative DNA damage. This effect could be attributed to the overexpression of
DYRK1A and
ETS2, which activate the DNA damage response in multiple cells, including stem cells, leading to a stem cell exhaustion phenotype. Mitotic instability related to the extra chromosome 21 contributes to further somatic aneuploidy and somatic mosaicism. Normally, these events would lead to increased tumor growth. However, unlike other constitutional aneuploidies such as Klinefelter and Turner syndromes, DS individuals seems to exhibit rare segmental chromosomal instability and somatic chromosomal translocations, translating into a lower susceptibility to solid tumors in both pediatric and adult populations
[26][43]. While there is debate regarding whether patients with Down syndrome exhibit a higher or lower rate of chromosomal instability compared to those without the condition, it is established that individuals with DS have a significantly lower incidence of prostate cancer. In cases where translocations involving the
ERG-
TMPRSS251 genes are common in the early phases of prostate cancer, it is noteworthy that such occurrences are less frequent among individuals with Down syndrome. Both of these genes are located on chromosome 21
[27][44].
Most studies concerning the protective effects of genes on chromosome 21 in DS have emphasized the concept that likely no single gene alone can exert a significant impact. This perspective takes into account that chromosome 21 also harbors a few genes known to have an oncogenic role or to stimulate cancer cell proliferation, growth, and metastasis
[7][24].
Among the various genes described, DYRK1A plays a significant role, seemingly implicated in all major phenotypic aspects of DS. For this reason, numerous biological approaches have been initiated to modulate the kinase activity of this protein. Interestingly, this approach will help to elucidate the multifaceted roles of this protein in the various aspects of cancer biology discovered thus far (Figure 1).
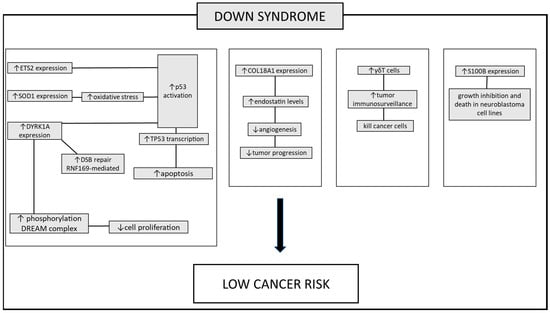
Figure 1. The pathways involved in protecting individuals with DS against tumors. Several mechanisms contributing to the lower incidence of tumors in these patients have been proposed. Increased TP53 apoptotic response: this is mediated by heightened expression of genes located on chromosome 21. These genes boost the transcription of TP53, ETS2, SOD1, and DYRK1A. Enhanced activation of DNA damage repair mechanisms leads to an increase in the repair of double-strand breaks, facilitated by elevated DYRK1A expression. The overexpression of the DYRK1A gene in DS patients leads to increased activation of the DREAM complex. This complex modulates the expression of genes involved in regulating the cell cycle, thereby reducing cell proliferation. Inhibition of tumor growth-promoting mechanisms: angiogenesis is suppressed due to heightened expression of COL18A1. Increased immune cell expression: the increased circulation of γδT cells, responsible for recognizing and suppressing tumor cells, plays a role in the lower tumor incidence. Additionally, the S100B protein, while contributing to the neurodegenerative process in DS patients, exhibits a contrasting effect in both human and murine neuroblastoma cell lines blocking tumor progression. Up and down arrows stand for “increased” or “decreased”.
1.2. Laron Syndrome
Laron syndrome (LS) is an autosomal recessive disorder caused by dysfunction in the growth hormone receptor. It is characterized by marked short stature, typical facial features, delayed sexual development, and obesity resulting from the inability to synthesize insulin-like growth factor I (IGF1) in response to growth hormone (GH)
[28][29][45,46]. In a study conducted by Laron et al., involving a cohort of 222 patients (comprising more than half of all known patients) with congenital IGF1 deficiency (169 of which had LS), none of these individuals had a history of cancer
[30][47]. In contrast, 9–24% of their family members had a malignancy history. This near-complete protection against cancer was observed in individuals who underwent GH treatment for a certain period, suggesting that the protective role started early during prenatal life and persisted into adulthood. Heterozygous carriers did not exhibit any dosage effect of this protective mechanism. The relationship between elevated levels of IGF1 and cancer is well established, elucidating higher cancer rates in obese individuals
[31][32][48,49]. Despite being obese, Laron patients remain free from cancer. Several studies on lymphoblastoid cell lines of LS revealed significant downregulation of genes including
CYCLINA1,
AKT3,
SP1,
SERPINB2,
VESICAN,
NPNT, and
OR5H2, while a few genes such as
UGT2B15,
UGT2B17, and
TXNIP, involved in xenobiotics detoxification and mitochondrial redox regulation, were upregulated
[33][50].
In vitro studies on lymphoblastoid cells from LS patients indicated reduced proliferation, altered cell cycle dynamics, decreased motility, increased apoptosis, resistance to oxidative stress challenges, and elevated expression of tumor suppressor genes. Notably, significantly reduced total and phosphorylated levels of IGF1R, a gene commonly overexpressed in various cancer types, were found in LS cells
[33][50]. This reduction correlated with parallel decreases in the phosphorylation of downstream signaling molecules AKT and ERK, which are typical mediators in the IGF1 and insulin pathways. The reduction in expression and activation of components within the IGF1R signaling axis might underlie the decrease in the mitogenic potential of LS cells.
Moreover, the distinct representation of IGF-binding proteins (IGFBPs) in LS-derived lymphoblasts could elucidate the lower tumor incidence in these patients. Specifically, mRNA levels of
IGFBP2,
IGFBP5, and
IGFBP6 were decreased in LS lymphoblastoids compared to healthy controls, while
IGFBP3 mRNA levels were increased in LS cells.
IGFBP3 has been recognized as an anti-oncogene in various tumors, consistent with its increased levels in LS.
IGFBP2 is typically associated with promoting tumorigenesis and T-cell proliferation, while
IGFBP5 and
IGFBP6 promote T-cell migration and act as a chemotactic agent for T-cells. Therefore, the reductions observed in these IGFBPs align with the protective role against cancer
[34][51].
The protective effect against cancer associated with a non-functioning insulin/IGF1 pathway was confirmed in
Caenorhabditis elegans. Mutations in the insulin/IGF-I like signaling pathway primarily increase lifespan. When this allele was combined with a null allele for a tumor suppressor gene causing germline tumors, a complete absence of tumor development was observed, suggesting that the lack of IGF1 effectively conferred resistance to tumors
[35][52]. Studies on mice treated with growth hormone receptor antagonists have demonstrated a lower incidence of carcinogenesis
[36][53].
An animal model of LS, the ‘Laron’ mouse, was created by disrupting the Gh receptor gene (
Ghr1). Similar to humans with LS, Laron mice exhibit low Igf1 and elevated Gh levels. Homozygous
Ghr1 knockout mice displayed the severe phenotype typical of Laron syndrome. However, heterozygous mice for the
Ghr1 axis showed minimal growth impairment, but presented an intermediate biochemical phenotype, with decreased Ghr- and Gh-binding protein expression and slightly reduced Igf1 levels
[37][54]. Transgenic mice expressing human GH and/or an agonist of the Igf1 receptor showed an increased incidence of breast tumor development
[38][55]. Conversely, mice carrying a transgene causing basal breast cancer in mice, when crossed with a tamoxifen-inducible conditional KO allele for the
Gh1r receptor, demonstrated that the ablation of the
Gh1r axis, after tumors reached a large size, completely inhibited the growth of breast cancer cells. These findings support the idea that growth hormone receptor antagonists may reduce the growth of cancer cells
[39][56].
Further investigations on the
Igf1r conditional KO mice aimed to elucidate the influential effects on cancer metastatic growth. Metastatic spread is a crucial aspect of cancer mortality. In one study, cells from Lewis lung carcinoma cells (LLC) were transplanted into mice with a
Igf1r KO background, meaning that the Igf1r axis was intact in cancer cells, but it was disrupted in the tumor microenvironment. The LLC, when transplanted into wild-type mice, led to multiple pulmonary metastases, while
Igf1r KO mice showed a reduced tumor burden. The same effect was replicated with a subcutaneous injection of a B16 melanoma cell line
[40][57]. These experiments in mice effectively demonstrated the potent inhibition of the Gh1r/Igf1 pathway, whether in cancer cells or in the supporting microenvironmental cells, leading to the inhibition of cancer cell growth, reduced inflammatory infiltration, enhanced apoptosis, reduced proliferation, and metastatic arrest. Ongoing trials with Gh1r/Igf1r antagonists will reveal their potential in this therapeutic approach, initiated from the clinical observation that LS patients do not develop cancer (
Figure 2).
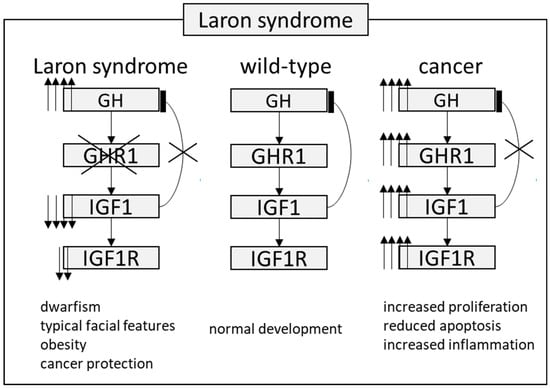
Figure 2. The pathway involved in protection against tumor in patients with Laron syndrome. During normal development, growth hormone (GH) is typically secreted by the pituitary gland and exerts its effects through the growth hormone receptor (GHR) present in various organs and cell types. Insulin-like growth factor 1 (IGF1) is released into the bloodstream, reaching different target organs and tissues. IGF1 negatively regulates GH secretion. However, in Laron syndrome, mutations in the GHR lead to increased GH levels, while IGF1 levels remain very low. This particular combination mediates a protective effect against cancer, despite these patients being obese. In cancer cases, a coordinated disruption of this axis at various levels results in markedly elevated IGF1 levels, thereby increasing downstream signaling. Up and down arrows stand for “increased” or “decreased”.
2. Reduced Cancer Risk in Syndromes with Dynamic Mutations
Fragile X syndrome (FXS) is caused by the absence in the brain of the fragile X mental retardation protein (FMRP). This protein is ubiquitously expressed suggesting that, in addition to its effects in brain, it may have fundamental roles in other organs
[41][58].
From the most comprehensive cumulative report of tumors in FXS obtained from 1988 to 2013, the rate of the most common malignancies in men (prostate, lung, and colorectal) were 6.8%, 4.5%, and 2.3%, respectively. That is significantly less than the expected estimated cancer incidence by site in the normal population (21%, 14%, and 8%, respectively). The male to female ratio was 1.58 to 1% and the female incidence of the three most common cancers (breast, lung, and colorectal) were 6.8%, 4.5%, and 2.3%, respectively, in comparison with the normal population (29%, 13%, and 8%)
[42][43][59,60].
There is evidence that FMRP expression can be linked to cancer. FMRP, as well as
FMR1 mRNA levels, correlate with prognostic indicators of aggressive breast cancer and lung metastasis
[44][61]. In particular, FMRP overexpression in murine breast primary tumors enhances lung metastasis, while its reduction has the opposite effect, regulating cell spreading and invasion. FMRP binds mRNAs involved in epithelial mesenchymal transition (EMT), often a prerequisite for metastases formation and invasion, including E-cadherin and Vimentin mRNAs, which are hallmarks of EMT and cancer progression
[44][61].
There is a statistically significant downregulation of the
WNT7A gene in FXS patients compared to healthy subjects; for this reason, the role of the WNT7A protein has also been extensively studied. Real-time PCR in FXS patients showed a real reduction in signal intensity in FXS males compared to healthy males
[45][62]. The reduced expression of the
WNT7A gene and its consequent downregulation of the β-catenin pathway may be related to a potential protection of FXS patients from cancer. Some of the target genes of this pathway, including the MYC, JUN, CYCLIND, and PPARδ genes, showed moderately reduced expression in FXS patients compared to normal subjects
[45][62].
Huntington’s disease (HD) is a progressive brain disorder caused by the pathological CAG expansion sequence in the
HTT gene. Patients with Huntington’s disease exhibit a significantly lower cancer incidence, up to 80 percent less than the general population
[46][63]. The analysis of data from 6540 subjects in the European Huntington’s disease network REGISTRY revealed a notably reduced age-standardized incidence rate, particularly evident in prostate and colorectal cancers, which exhibited the lowest rates
[46][63]. Several potential factors may account for these lower cancer rates. Factors such as lower life expectancy might contribute, and there could be instances where cancer is underdiagnosed among individuals with HD, especially in later stages of the illness. This could occur as relevant signs or symptoms may be overlooked or overshadowed by HD symptoms, such as cachexia.
Notably, lung cancer was the only cancer found to occur as frequently in the HD population as in the general population, as reported by Turner et al.
[47][64]. They attributed this finding to the higher rate of smoking among individuals with HD, a trend observed in various other psychotic conditions as well
[48][65].
While the incidence of cancer is lower in HD patients than in age-matched controls, HD-causing CAG expansions of
HTT accelerate the progression of breast tumors and the development of metastases in mouse models of breast cancer. In particular, Thion et al.
[49][66] showed that the length of
HTT CAG correlates with a lower incidence of ovarian cancer in carriers of the
BRCA2 mutation and that CAG repeat length in the long
HTT allele can be a factor in metastasis in sporadic breast cancer (HER+ subtype)
[49][66]. One of the possible mechanisms linking HD and cancer protection may be RNA toxicity. Murmann and colleagues constructed small interfering RNAs based on
HTT CAG repeats; these siRNAs induce cell death in vitro in all tested cancer cell lines and slow down tumor growth in a preclinical mouse model of ovarian cancer, with no signs of toxicity to the mice
[50][67]. To indirectly confirm the role of RNA toxicity, in other neurodegenerative conditions with polyglutamine expansions a lower cancer incidence was also observed (
Figure 3).
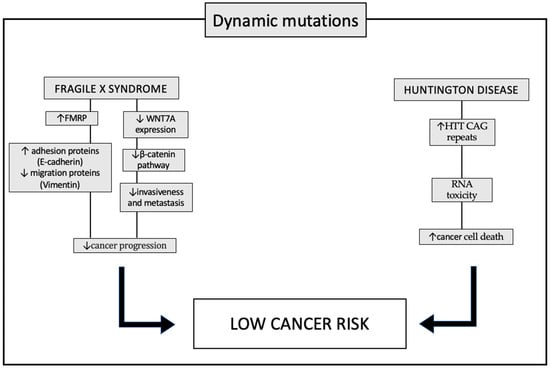
Figure 3. Pathways involved in protection against tumors in patient with dynamic mutations: fragile X syndrome (FXS) and Huntington’s disease (HD). In patients with FXS, increased levels of the FMRP (fragile X mental retardation 1 protein) appear to alter the expression of proteins that promote tumor progression. In particular, the typical invasiveness of tumor cells is inhibited through the decreased expression of the vimentin protein, and cell adhesion is instead promoted, thanks to the increased expression of E-cadherin. Another mechanism underlying the lower incidence of cancer in patients with FXS is the decreased expression of the WNT7A (WNT family member 7A) gene which, through downregulation of the β-catenin pathway, prevents the invasiveness of tumor cells. In patients with HD, however, one of the molecular causes supposed at the basis of the lower incidence of tumors is the increased level of RNAs, which have a toxic action, not only towards neuronal cells, causing the typical clinical signs of the disease, but also to tumor cells. Up and down arrows stand for “increased” or “decreased”.