Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jason Zhu and Version 1 by Manju Tewari.
P2X receptors are a family of seven ATP-gated ion channels that trigger physiological and pathophysiological responses in various cells. Five of the family members are sensitive to low concentrations of extracellular ATP, while the P2X6 receptor has an unknown affinity. The last subtype, the P2X7 receptor, is unique in requiring millimolar concentrations to activate in humans fully. This low sensitivity imparts the agonist with the ability to act as a damage-associated molecular pattern that triggers the innate immune response in response to the elevated extracellular ATP levels accompanying inflammation and tissue damage.
- ATP
- microglia
- purinergic receptors
- P2X7
- innate immunity
- tumor microenvironment
1. Introduction
Microglia show constitutive expression of p2rx4 and p2rx7 in both rodents and humans [1][2]. Microglial P2X4Rs play critical roles in rodent models of neuropathic pain [3][4], experimental allergic encephalomyelitis [5][6], and chronic migraine [7]. In keeping with these studies, relatively low concentrations of eATP (≤100 µM) evoke desensitizing non-selective inward currents and sustained outward K+ currents in murine microglia that most likely result from activation of P2X4Rs and P2Y12Rs, respectively [8][9][10][11][12][13]. Surprisingly, these same concentrations of eATP fail to trigger inward membrane current or Ca2+ influx in cultured human microglia, suggesting that either the p2rx4 is downregulated in culture, the mRNA is not translated, or the properly translated protein is not trafficked to the cell surface membrane [14][15].
In contrast, and as expected from work on mice [16], short applications of higher concentrations of eATP (≥1 mM) or the higher affinity ATP analog, 2′,3′-O-(benzoyl-4benzoyl)-ATP (BzATP), evoke inward currents in human microglia with properties expected of a P2X7R-mediated response (Figure 1); the current is cation non-selective, carried in part by Ca2+, facilitated during prolonged exposure to agonists, and blocked by P2X7R antagonists [14]. The resting membrane potential of human microglia is unknown. In rodents, it varies with age [17] but averages around −40 mV [9][18]. At this potential, eATP activation of P2X7Rs causes Na+ and Ca2+ to rush into the cell as K+ exits. The net result is membrane depolarization. In rodents, the inward Ca2+ current increases the concentration of free intracellular Ca2+ ([Ca2+]i), which triggers cell cycle progression [19], the release of TNF-α [20] and plasminogen [21], activation of the transcription factor NFAT [22], disruption of the cytoskeleton [23], and the production of H2O2 [24]. At the same time, the outward K+ current decreases the concentration of intracellular K+ ([K+]i), leading to activation of the NLRP3 inflammasome and maturation and release of the proinflammatory cytokines, IL-1β and IL-18 [25]. While it is well accepted that the inflammasome activates when [K+]i drops below 90 mM [26], more recent evidence suggests that the P2X7R is not the primary K+ efflux pathway [27]. Rather, the two-pore K+ channels, THIK-1 and TWIK-2, are responsible in microglia [9][28][29] and macrophages [30], respectively. The data supporting a role for THIK-1 and TWIK-2 are convincing, and the conclusions are firm. However, it is unclear why K+ efflux through the P2X7R is not sufficient by itself. In the microglial study of Madry et al., the simple fact that 2 mM ATP did not evoke a P2X7R-mediated current is enough to eliminate this receptor from consideration [9]. The lack of response is surprising because others report robust eATP-gated currents with properties unique to P2X7Rs in murine and human microglia [14][16][31][32], perhaps suggesting that P2X7R expression resembles P2X4R expression in its sensitivity to the choice of animal, the activation state of the microglia, and/or the experimental protocols used to study the cell. Regardless, when present, activation of P2X7Rs results in a large efflux of 86Rb+, a proxy for K+, in J774 macrophages and presumably in microglia [33]. Further, eATP promotes recruitment and colocalization of microglial P2X7Rs and NLRP3 to discrete sites of the subplasmalemmal cytoplasm, suggesting that the inflammasome is positioned close enough to directly sense the P2X7R-mediated local drop in [K+]i [34].
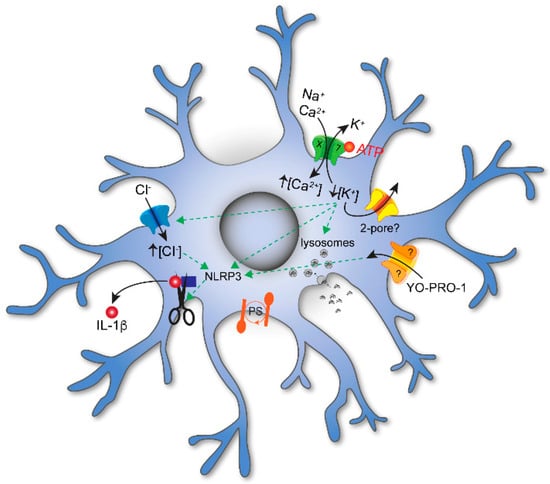
Figure 1. P2X signaling in microglia. Extracellular ATP evokes influx of Na+ and Ca2+ and efflux of K+ through P2X7Rs embedded in the plasmalemmal membrane. ATP-gated efflux of K+ is also thought to involve additional channels; at present, the identity of these channels is unknown but may involve 2–pore K+ channels. The decrease in [K+]i results in influx of Cl− and the maturation and release of the proinflammatory cytokine IL–1β. Activation of P2X7Rs also increases membrane permeabilization of large organic cations like YO-PRO1, decreases phagocytosis, and initiates phosphatidylserine (PS) exposure on the cell surface. While YO-PRO1 permeates the P2X7R pore, other pathways (marked with a “?”) are also thought to play a role.
2. Membrane Permeabilization and Cell Lysis
Applications of eATP that last longer than a few seconds result in membrane permeabilization, a hallmark property of P2X7R activation [35][36][37]. Permeabilization is the process by which eATP triggers membrane transport of hydrophilic solutes with molecular masses of <900 Da in a direction determined by their electrochemical potential. The process is reversible [38] and does not necessarily lead to cell death. The ability of eATP to permeabilize membranes was first discovered in mouse 3T3 fibroblasts [39], rat mast cells [40], and mouse J774 macrophages [33][41][42], and later described in mouse microglia [43]. Originally thought to be a unique property of P2X7Rs, it was subsequently shown to accompany the activation of purinergic P2X2Rs, P2X3Rs and P2X4Rs [44][45][46] as well as proton-gated TRPV1 receptors [47]. While the physiological consequence of membrane permeabilization is the loss of cytoplasmic components such as ATP, cGMP, glutamate, and spermidine, the underlying process is typically measured as uptake of polyatomic fluorescent biomarkers such as the nucleic acid stains ethidium bromide and YO-PRO-1 [36][37]. For example, in HEK293 cells expressing recombinant P2X7Rs, BzATP triggers an increase in fluorescence that develops slowly over the course of seconds to minutes as dye enters the cell and intercalates DNA [35]. The primary route of entry is unclear [36]. That some of the dye transits the P2X7R pore is convincingly proven using reconstituted receptors and liposomes that lack other proteins [48]. At the same time, eATP and P2X7Rs may activate secondary transport pathways. For example, pannexin-1 is a plasma membrane cation-selective channel related to gap junction proteins that shows a close membrane association with P2X7Rs. Pharmacologically inhibiting pannexin-1 blocks the initial phase of eATP-gated ethidium uptake in human lung alveolar macrophages, demonstrating that it is partially responsible for membrane permeabilization to cationic macromolecules in these cells [49]. The same drugs also block P2X7R-dependent release of IL-1β, suggesting that pannexin-1 is a critical component of eATP-mediated NLRP3 inflammasome activation [49][50]. In contrast, blocking pannexin-1 has no effect on eATP-mediated membrane permeabilization of monocyte-derived human macrophages [51] or cultured human microglia [14], suggesting that local paracrine signaling active in tissue microenvironments may recruit distinct permeabilization pathways in a tissue-specific manner. The hypothesis that eATP gates multiple transport portals is further supported by the finding that, in some cases, P2X7Rs increase membrane permeability to both cations (YO-PRO-1 and ethidium) and anions (glutamate and Lucifer yellow) in the same cell [42][52][53][54]. It is unlikely that large anions travel through P2X7Rs because these channels show a strong preference for cations [35]. In addition, the uptake of cations is temperature-dependent, whereas the uptake of anions is not [55], again supporting the presence of multiple permeation pathways in some cells. With specific regard to human microglia, eATP does not trigger uptake of large anions in cultured cells [14], although this may reflect the downregulation of genes encoding the necessary pathway; additional experiments on cells that more closely resemble the natural phenotype of in situ microglia are needed.
The physiological and pathophysiological outcomes of membrane permeabilization are uncertain [36] and have not been extensively investigated in microglia. As mentioned above, the process is reversible and does not necessarily lead to cell death. For example, a 30 min application of ATP or BzATP causes significant uptake of cationic YO-PRO-1 in human microglia without inducing the release of lactate dehydrogenase, demonstrating that permeabilization is not lytic under these conditions [14]. In contrast, longer incubations result in necrosis or apoptosis in murine microglia, a result that is prevented by pre-incubation of a P2X7R antagonist [56][57]. Cell lysis is blocked by P2X7R antagonists and absent in cells isolated from P2X7R−/− animals, suggesting that the activation of P2X7R is a critical component of the lytic pathway [2]. Interestingly, eATP-activation of P2X2Rs permeabilizes membranes but does not kill cells, suggesting that permeabilization and lysis proceed through different intracellular signaling pathways [45]. If cell death is not the ultimate consequence, then what purpose does permeabilization serve? The recent work of the Grutter laboratory suggests one possibility [46]. Spermidine is a naturally occurring intracellular polyamine that acts at extracellular sites to allosterically modulate ion channel gating [58]. To be effective, it must be secreted. Spermidine permeates multiple subtypes of P2XRs, including P2X7Rs, suggesting that these receptors represent an eATP-dependent egress pathway for polyamines, an effect that may help to explain the ability of P2X7Rs to modulate the activity of neighboring ion channels [37].
3. Membrane Blebbing and Microvesicular Shedding
Non-apoptotic membrane blebbing occurs when the actin cytoskeleton separates from the plasma membrane; detachment allows the hydrostatic pressure within the cell to push the membrane through the actin cortex, forming an outwardly facing membrane extrusion [59][60]. eATP, working through P2X7Rs, is a potent initiator of non-apoptotic membrane blebbing in many cell types, including human macrophages [51] and murine microglia [61][62][63]. Here, blebbing occurs within minutes of P2X7R activation [62] and reverses when the agonist is removed [51]. As is the case of membrane permeabilization (see above), the physiological sequela of membrane blebbing is uncertain. In tumor cells, blebs facilitate cytokinesis [64], and in streptolysin-permeabilized human embryonic kidney cells, they trap damaged membrane segments and limit further cellular damage [65]. In THP-1 monocytes [62] and primary mouse microglia [61], bleb formation provides a vehicle for IL-1β release. Here, eATP activation of P2X7Rs results in rapid movement of phosphatidylserine and acid sphingomyelinase to the outer leaflet of the plasma membrane, resulting in the formation of small (40–80 nm) membrane-derived microvesicles that contain IL-1β. Surprisingly, eATP does not cause blebbing in cultured human microglia [14].
4. Cytokines and Reactive Oxygen Species (ROS)
The ability of eATP acting on purinergic P2 receptors to function as a DAMP has long established a role for eATP in regulating neuroinflammatory immune responses [66][67]. The immune response is characterized by a proinflammatory state, which is driven by immune cell activation and the subsequent release of proinflammatory cytokines and reactive oxygen species (ROS) [24][68][69]. In the presence of CNS injury, infection, or neurodegeneration, copious amounts of ATP are released into the extracellular environment from stressed and dying cells [70]. At high enough concentrations, this eATP can promote macrophage and microglial activation, which drives a cascade of P2X7R-mediated events that concludes with the time-dependent release of proinflammatory cytokines, including IL-6, IL-18, TNF-α, and IL-1β [14][69][71][72][73]. Specifically, P2X7R-mediated IL-1β release occurs through a multi-step process that requires priming first by cellular stress or pathogens (i.e., LPS) to stimulate the production of immature pro-IL-1β [25][74][75]. Upon accumulation in the cytosol, pro-IL-1β requires a secondary hit to promote its maturation and secretion driven by caspase-1 activity. Importantly, cells within a quiescent state store caspase-1 in an inactive form (pro-caspase-1), which requires cellular stimulation via DAMPs to undergo maturation to the active form [25]. In the case of eATP functioning as a DAMP, P2X7R activation drives significant K+ efflux from the intracellular environment, which subsequently promotes NLRP3 inflammasome complex formation [67][70][75]. The NLRP3 inflammasome is composed of a primary scaffold protein (NLRP3) that recruits the accessory protein ASC, which mediates pro-caspase-1 recruitment and activation [25][67]. Upon activation, caspase-1 drives the cleavage of pro-IL-1β into its active form, which can subsequently be secreted from the cell upon microvesicle shedding from the plasma membrane [61][62]. Importantly, P2X7R activation in microglia also promotes reactive oxygen species (ROS) formation [24][76]. Upon stimulation, P2X7R drives ROS production via p38 MAPK-dependent NADPH oxidase activation [24]. Notably, both the P2X7R-mediated release of proinflammatory cytokines and ROS are highly characterized in the pathophysiology of neurodegenerative diseases [70]. In Alzheimer’s Disease, the P2X7R is significantly upregulated adjacent to the characteristic Aβ plaques where surrounding activated microglial populations are colocalized [24]. Importantly, Aβ stimulates microglial activation, thereby driving the release of proinflammatory cytokines and ROS, which induce pro-apoptotic gene activity, thus mediating the death of neuronal cell populations and exacerbating neuroinflammation [70][77].
5. Tumor Microenvironment
Tumorigenesis frequently occurs at chronically inflamed tissue sites [78][79][80], where the rate at which the tumor proliferates is largely dependent on a delicate balance of immunosuppressive and immunostimulating cell types coexisting within the tumor microenvironment (TME) [5][81][82][83][84][85]. Underlying components of the TME include stromal cells, fibroblasts, endothelial cells, and infiltrating innate (TAMs, myeloid-derived suppressor cells, dendritic cells) and adaptive (T cells) immune cells [78][86]. Communication between cells occurs through the release of growth factors (VEGF), cytokines (IL-6), chemokines, components of the extracellular matrix, and purines to dictate tumor growth [78][80][87]. Tumor cells express P2X7Rs that drive proliferation by enhancing cellular metabolism and angiogenesis when the concentration of eATP is relatively low [84][88][89]. In contrast, when eATP rises to concentrations ≥100 µM as the result of hypoxic tissue necrosis, eATP promotes tumor cell cytotoxicity through sustained membrane permeabilization [81][84][90]. The ability of ATP to kill cancer cells justifies the development of selective P2X7R agonists as a therapeutic cancer target [90][91].
The immune cells that infiltrate tumors also express high densities of P2X7Rs, which in part determine whether these cells work to promote or eliminate tumor cells [79][92]. P2X7R-driven NLRP3 inflammasome complex activation and subsequent IL-1β release largely account for the immunostimulating qualities of P2X7R. Namely, IL-1β secretion from dendritic cells primes antigen-specific CD8+ T-cells, which release IFN-γ to exert their antitumor effects [79][93]. This tumor-eradicating role is exemplified in cancer models utilizing P2X7R-deficient mice (p2rx7−/−). In the absence of P2X7R, inoculated tumors both proliferated and metastasized at a faster rate compared to those brought up in wild-type mice (p2rx7+/+) [94]. Conversely, P2X7R activation on myeloid-derived suppressor cells fosters tumor-promotion upon the production and release of immunosuppressive factors, including reactive oxygen species, arginase-1, and TGF-β1 [79][87]. Additionally, P2X7R upregulation in glioma-associated microglia drives immunosuppression upon facilitating NLRP3 inflammasome activation and IL-1β release [92][95]. Proinflammatory IL-1β stimulates glioma cells to produce TGF-β, which mediates subsequent upregulation of VEGF, leading to tumor proliferation via increased angiogenesis [96][97][98]. Importantly, eATP also exhibits immunosuppressive effects upon its breakdown to adenosine via ectonucleotidases CD39 and CD73. Particularly, Tregs’ characteristic immunosuppressive activity is based on its high expression level of both ectonucleotidases. Free adenosine can then target P1 A2ARs to inhibit tumor-infiltrating cytotoxic CD8+ T cells [90][97].
6. Cell Death and Disease
Sustained stimulation with ATP is a potent catalytic stimulus for several cell types, including microglial cells, and the available literature clearly point towards the involvement of P2X7R in ATP-induced cell death, as reviewed by Peter Illes [99]. P2X7R has been described as a death receptor [56][100]. Short periods of P2X7R activation are cytotoxic, and once activated, the P2X7R sets in motion an irreversible death process [101][102]. Cells primed with inflammatory mediators (e.g., lipopolysaccharide) are particularly susceptible to the toxic actions of ATP [103], and this priming effect may alter the distribution or activation of P2X7 receptors in cell membranes [104].
Studies in culture strongly suggest a role for P2X7R-mediated cell death in a number of neurodegenerative diseases. Specific examples include rat microglial cell lines N9 and N13 [57], murine microglial cell line EOC13 [105], mouse primary microglia [24][102], and enteric glia [106].
7. Oxygen Glucose Deprivation
Oxygen glucose deprivation (OGD) is often used to study ischemic cell death. It negatively impacts microglia motility and induces microglia cell death. Upregulation of the P2X4R and P2X7R is reported to occur in N9 microglial cells deprived of oxygen [107]. Further, the same study suggests that metabolic stress like OGD induces massive release of extracellular ATP, which in turn activates cortical P2X and P2Y receptors. Several P2 receptors (P2X1R, P2X2R, P2X3R, P2X5R, and P2Y11R) alter the homeostatic balance of Ca2+ and Na+ fluxes, triggering both necrotic and apoptotic pathways [108]. In a similar fashion, P2X4 and P2X7 receptors induce the microglial release of proinflammatory cytokines [68] and subsequent neuronal death. Blocking the receptors with the P2 antagonists PPADS and TNP-ATP reduced microglia activation and rescued cortical cells from OGD-induced cell death [107]. OGD-induced microglial cell death has also been studied in BV2 cells, where the pharmacological inhibition of P2X7R using Brilliant Blue G (BBG) significantly reduced OGD-induced BV2 cell death. Similar results were observed in neonatal hippocampal slices. Here, OGD increases extracellular ATP, and treatments that decreased the concentration of extracellular ATP or reduced the availability of P2X7R receptors inhibited OGD-induced microglia cell death [109]. The depletion of extracellular Ca2+ also significantly inhibits cell death, indicating that OGD induces Ca2+-dependent microglia cell death [109]. Further in vivo studies performed on a middle cerebral artery occlusion rat model showed that inhibition of P2X7R expression by promoting degradation of ATP protects against the brain injury produced by OGD [110]. However, P2X7Rs are not the sole contributors to the purine- and calcium-dependent ischemic cell death and other mechanisms remain to be discovered.
References
- Illes, P.; Rubini, P.; Ulrich, H.; Zhao, Y.; Tang, Y. Regulation of Microglial Functions by Purinergic Mechanisms in the Healthy and Diseased CNS. Cells 2020, 9, 1108.
- He, Y.; Taylor, N.; Fourgeaud, L.; Bhattacharya, A. The Role of Microglial P2X7: Modulation of Cell Death and Cytokine Release. J. Neuroinflamm. 2017, 14, 135.
- Inoue, K.; Tsuda, M. Microglia in Neuropathic Pain: Cellular and Molecular Mechanisms and Therapeutic Potential. Nat. Rev. Neurosci. 2018, 19, 138–152.
- Jurga, A.M.; Piotrowska, A.; Makuch, W.; Przewlocka, B.; Mika, J. Blockade of P2X4 Receptors Inhibits Neuropathic Pain-Related Behavior by Preventing MMP-9 Activation and, Consequently, Pronociceptive Interleukin Release in a Rat Model. Front. Pharmacol. 2017, 8, 48.
- Di Virgilio, F.; Sarti, A.C. Microglia P2X4 Receptors as Pharmacological Targets for Demyelinating Diseases. EMBO Mol. Med. 2018, 10, e9369.
- Zabala, A.; Vazquez-Villoldo, N.; Rissiek, B.; Gejo, J.; Martin, A.; Palomino, A.; Perez-Samartín, A.; Pulagam, K.R.; Lukowiak, M.; Capetillo-Zarate, E.; et al. P2X4 Receptor Controls Microglia Activation and Favors Remyelination in Autoimmune Encephalitis. EMBO Mol. Med. 2018, 10, e8743.
- Long, T.; He, W.; Pan, Q.; Zhang, S.; Zhang, Y.; Liu, C.; Liu, Q.; Qin, G.; Chen, L.; Zhou, J. Microglia P2X4 Receptor Contributes to Central Sensitization Following Recurrent Nitroglycerin Stimulation. J. Neuroinflamm. 2018, 15, 245.
- Swiatkowski, P.; Murugan, M.; Eyo, U.; Wang, Y.; Rangaraju, S.; Oh, S.; Wu, L.-J. Activation of Microglial P2Y12 Receptor Is Required for Outward Potassium Currents in Response to Neuronal Injury. Neuroscience 2016, 318, 22–33.
- Madry, C.; Kyrargyri, V.; Arancibia-Cárcamo, I.L.; Jolivet, R.; Kohsaka, S.; Bryan, R.M.; Attwell, D. Microglial Ramification, Surveillance, and Interleukin-1β Release Are Regulated by the Two-Pore Domain K+ Channel THIK-1. Neuron 2018, 97, 299–312.e6.
- Nörenberg, W.; Langosch, J.; Gebicke-Haerter, P.; Illes, P. Characterization and Possible Function of Adenosine 5′-Triphosphate Receptors in Activated Rat Microglia. Br. J. Pharmacol. 1994, 111, 942–950.
- Walz, W.; Ilschner, S.; Ohlemeyer, C.; Banati, R.; Kettenmann, H. Extracellular ATP Activates a Cation Conductance and a K+ Conductance in Cultured Microglial Cells from Mouse Brain. J. Neurosci. 1993, 13, 4403–4411.
- Illes, P.; Nörenberg, W.; Gebicke-Haerter, P.J. Molecular mechanisms of microglial activation. B. Voltage- and purinoceptor-operated channels in microglia. Neurochem. Int. 1996, 29, 13–24.
- Toulme, E.; Garcia, A.; Samways, D.; Egan, T.M.; Carson, M.J.; Khakh, B.S. P2X4 Receptors in Activated C8-B4 Cells of Cerebellar Microglial Origin. J. Gen. Physiol. 2010, 135, 333–353.
- Janks, L.; Sharma, C.V.R.; Egan, T.M. A Central Role for P2X7 Receptors in Human Microglia. J. Neuroinflamm. 2018, 15, 325.
- McLarnon, J.G. Purinergic Mediated Changes in Ca2+ Mobilization and Functional Responses in Microglia: Effects of Low Levels of ATP. J. Neurosci. Res. 2005, 81, 349–356.
- Haas, S.; Brockhaus, J.; Verkhratsky, A.; Kettenmann, H. ATP-Induced Membrane Currents in Ameboid Microglia Acutely Isolated from Mouse Brain Slices. Neuroscience 1996, 75, 257–261.
- Schilling, T.; Eder, C. Microglial K+ Channel Expression in Young Adult and Aged Mice. Glia 2015, 63, 664–672.
- Nörenberg, W.; Gebicke-Haerter, P.J.; Illes, P. Voltage-Dependent Potassium Channels in Activated Rat Microglia. J. Physiol. 1994, 475, 15–32.
- Bianco, F.; Ceruti, S.; Colombo, A.; Fumagalli, M.; Ferrari, D.; Pizzirani, C.; Matteoli, M.; Di Virgilio, F.; Abbracchio, M.P.; Verderio, C. A Role for P2X7 in Microglial Proliferation. J. Neurochem. 2006, 99, 745–758.
- Hide, I.; Tanaka, M.; Inoue, A.; Nakajima, K.; Kohsaka, S.; Inoue, K.; Nakata, Y. Extracellular ATP Triggers Tumor Necrosis Factor-α Release from Rat Microglia. J. Neurochem. 2000, 75, 965–972.
- Inoue, K.; Nakajima, K.; Morimoto, T.; Kikuchi, Y.; Koizumi, S.; Illes, P.; Kohsaka, S. ATP stimulation of Ca2+-dependent plasminogen release from cultured microglia. Br. J. Pharmacol. 1998, 123, 1304–1310.
- Ferrari, D.; Stroh, C.; Schulze-Osthoff, K. P2X7/P2Z Purinoreceptor-mediated Activation of Transcription Factor NFAT in Microglial Cells. J. Biol. Chem. 1999, 274, 13205–13210.
- Mackenzie, A.B.; Young, M.T.; Adinolfi, E.; Surprenant, A. Pseudoapoptosis Induced by Brief Activation of ATP-gated P2X7 Receptors. J. Biol. Chem. 2005, 280, 33968–33976.
- Parvathenani, L.K.; Tertyshnikova, S.; Greco, C.R.; Roberts, S.B.; Robertson, B.; Posmantur, R. P2X7 Mediates Superoxide Production in Primary Microglia and Is Up-Regulated in a Transgenic Mouse Model of Alzheimer’s Disease. J. Biol. Chem. 2003, 278, 13309–13317.
- Di Virgilio, F. Liaisons Dangereuses: P2X7 and the Inflammasome. Trends Pharmacol. Sci. 2007, 28, 465–472.
- Pétrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 Inflammasome is Triggered by Low Intracellular Potassium Concentration. Cell Death Differ. 2007, 14, 1583–1589.
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489.
- Drinkall, S.; Lawrence, C.B.; Ossola, B.; Russell, S.; Bender, C.; Brice, N.B.; Dawson, L.A.; Harte, M.; Brough, D. The Two Pore potassium Channel THIK-1 Regulates NLRP3 Inflammasome Activation. Glia 2022, 70, 1301–1316.
- Ossola, B.; Rifat, A.; Rowland, A.; Hunter, H.; Drinkall, S.; Bender, C.; Hamlischer, M.; Teall, M.; Burley, R.; Barker, D.F.; et al. Characterisation of C101248: A Novel Selective THIK-1 Channel Inhibitor for the Modulation of Microglial NLRP3-Inflammasome. Neuropharmacology 2023, 224, 109330.
- Di, A.; Xiong, S.; Ye, Z.; Malireddi, R.S.; Kometani, S.; Zhong, M.; Mittal, M.; Hong, Z.; Kanneganti, T.-D.; Rehman, J.; et al. The TWIK2 Potassium Efflux Channel in Macrophages Mediates NLRP3 Inflammasome-Induced Inflammation. Immunity 2018, 49, 56–65.e4.
- Chessell, I.P.; Michel, A.D.; Humphrey, P.P.A. Properties of the Pore-Forming P2x7 Purinoceptor in Mouse NTW8 Microglial Cells. Br. J. Pharmacol. 1997, 121, 1429–1437.
- Raouf, R.; Chabot-Doré, A.-J.; Ase, A.R.; Blais, D.; Séguéla, P. Differential Regulation of Microglial P2X4 and P2X7 ATP Receptors following LPS-Induced Activation. Neuropharmacology 2007, 53, 496–504.
- Steinberg, T.; Silverstein, S. Extracellular ATP4-Promotes Cation Fluxes in the J774 Mouse Macrophage Cell Line. J. Biol. Chem. 1987, 262, 3118–3122.
- Franceschini, A.; Capece, M.; Chiozzi, P.; Falzoni, S.; Sanz, J.M.; Sarti, A.C.; Bonora, M.; Pinton, P.; Di Virgilio, F. The P2X7 Receptor Directly Interacts with the NLRP3 Inflammasome Scaffold Protein. FASEB J. 2015, 29, 2450–2461.
- Virginio, C.; MacKenzie, A.; North, R.A.; Surprenant, A. Kinetics of Cell Lysis, Dye Uptake and Permeability Changes in Cells Expressing the Rat P2X7 Receptor. J. Physiol. 1999, 519, 335–346.
- Di Virgilio, F.; Schmalzing, G.; Markwardt, F. The Elusive P2X7 Macropore. Trends Cell Biol. 2018, 28, 392–404.
- Peverini, L.; Beudez, J.; Dunning, K.; Chataigneau, T.; Grutter, T. New Insights Into Permeation of Large Cations Through ATP-Gated P2X Receptors. Front. Mol. Neurosci. 2018, 11, 265.
- Surprenant, A.; Rassendren, F.; Kawashima, E.; North, R.A.; Buell, G. The Cytolytic P2Z Receptor for Extracellular ATP Identified as a P2X Receptor (P2X7). Science 1996, 272, 735–738.
- Rozengurt, E.; Heppel, L.A. A Specific Effect of External ATP on the Permeability of Transformed 3T3 Cells. Biochem. Biophys. Res. Commun. 1975, 67, 1581–1588.
- Cockcroft, S.; Gomperts, B.D. ATP Induces Nucleotide Permeability in Rat Mast Cells. Nature 1979, 279, 541–542.
- Steinberg, T.H.; Newman, A.S.; Swanson, J.A.; Silverstein, S.C. ATP4-Permeabilizes the Plasma Membrane of Mouse Macrophages to Fluorescent Dyes. J. Biol. Chem. 1987, 262, 8884–8888.
- Buisman, H.P.; Steinberg, T.H.; Fischbarg, J.; Silverstein, S.C.; Vogelzang, S.A.; Ince, C.; Ypey, D.L.; Leijh, P.C. Extracellular ATP Induces a Large Nonselective Conductance in Macrophage Plasma Membranes. Proc. Natl. Acad. Sci. USA 1988, 85, 7988–7992.
- Ferrari, D.; Villalba, M.; Chiozzi, P.; Falzoni, S.; Ricciardi-Castagnoli, P.; Di Virgilio, F. Mouse Microglial Cells Express a Plasma Membrane Pore Gated by Extracellular ATP. J. Immunol. 1996, 156, 1531–1539.
- Khakh, B.S.; Bao, X.R.; Labarca, C.; Lester, H.A. Neuronal P2X Transmitter-Gated Cation Channels Change Their Ion Selectivity in Seconds. Nat. Neurosci. 1999, 2, 322–330.
- Virginio, C.; MacKenzie, A.; Rassendren, F.A.; North, R.A.; Surprenant, A. Pore Dilation of Neuronal P2X Receptor Channels. Nat. Neurosci. 1999, 2, 315–321.
- Harkat, M.; Peverini, L.; Cerdan, A.H.; Dunning, K.; Beudez, J.; Martz, A.; Calimet, N.; Specht, A.; Cecchini, M.; Chataigneau, T.; et al. On the Permeation of Large Organic Cations through the Pore of ATP-Gated P2X Receptors. Proc. Natl. Acad. Sci. USA 2017, 114, E3786–E3795.
- Chung, M.-K.; Güler, A.D.; Caterina, M.J. TRPV1 Shows Dynamic Ionic Selectivity during Agonist Stimulation. Nat. Neurosci. 2008, 11, 555–564.
- Karasawa, A.; Michalski, K.; Mikhelzon, P.; Kawate, T. The P2X7 Receptor Forms a Dye-Permeable Pore Independent of Its Intracellular Domain but Dependent on Membrane Lipid Composition. eLife 2017, 6, e31186.
- Pelegrin, P.; Surprenant, A. Pannexin-1 Mediates Large Pore Formation and Interleukin-1β Release by the ATP-Gated P2X7 Receptor. EMBO J. 2006, 25, 5071–5082.
- Pelegrin, P.; Surprenant, A. The P2X7 Receptor–Pannexin Connection to Dye Uptake and IL-1β Release. Purinergic Signal. 2009, 5, 129–137.
- Janks, L.; Sprague, R.S.; Egan, T.M. ATP-Gated P2X7 Receptors Require Chloride Channels to Promote Inflammation in Human Macrophages. J. Immunol. 2019, 202, 883–898.
- Duan, S.; Anderson, C.M.; Keung, E.C.; Chen, Y.; Chen, Y.; Swanson, R.A. P2X7 Receptor-Mediated Release of Excitatory Amino Acids from Astrocytes. J. Neurosci. 2003, 23, 1320–1328.
- Marques-Da-Silva, C.; Chaves, M.M.; Rodrigues, J.C.; Corte-Real, S.; Coutinho-Silva, R.; Persechini, P.M. Differential Modulation of ATP-Induced P2X7-Associated Permeabilities to Cations and Anions of Macrophages by Infection with Leishmania amazonensis. PLoS ONE 2011, 6, e25356.
- Ugur, M.; Ugur, Ö. A Mechanism-Based Approach to P2X7 Receptor Action. Mol. Pharmacol. 2019, 95, 442–450.
- Schachter, J.; Motta, A.P.; Zamorano, A.d.S.; da Silva-Souza, H.A.; Guimarães, M.Z.P.; Persechini, P.M. ATP-Induced P2X7-Associated Uptake of Large Molecules Involves Distinct Mechanisms for Cations and Anions in Macrophages. J. Cell Sci. 2008, 121, 3261–3270.
- Di Virgilio, F. ATP as a Death Factor. BioFactors 1998, 8, 301–303.
- Ferrari, D.; Chiozzi, P.; Falzoni, S.; Susino, M.D.; Collo, G.; Buell, G.; Di Virgilio, F. ATP-Mediated Cytotoxicity in Microglial Cells. Neuropharmacology 1997, 36, 1295–1301.
- Guerra, G.P.; Rubin, M.A.; Mello, C.F. Modulation of Learning and Memory by Natural Polyamines. Pharmacol. Res. 2016, 112, 99–118.
- Charras, G.T.; Coughlin, M.; Mitchison, T.J.; Mahadevan, L. Life and Times of a Cellular Bleb. Biophys. J. 2008, 94, 1836–1853.
- Weng, N.J.-H.; Talbot, P. The P2X7 Receptor is an Upstream Regulator of Dynamic Blebbing and a Pluripotency Marker in Human Embryonic Stem Cells. Stem Cell Res. 2017, 23, 39–49.
- Bianco, F.; Pravettoni, E.; Colombo, A.; Schenk, U.; Möller, T.; Matteoli, M.; Verderio, C. Astrocyte-Derived ATP Induces Vesicle Shedding and IL-1β Release from Microglia. J. Immunol. 2005, 174, 7268–7277.
- MacKenzie, A.; Wilson, H.L.; Kiss-Toth, E.; Dower, S.K.; North, R.A.; Surprenant, A. Rapid Secretion of Interleukin-1β by Microvesicle Shedding. Immunity 2001, 15, 825–835.
- Bianco, F.; Perrotta, C.; Novellino, L.; Francolini, M.; Riganti, L.; Menna, E.; Saglietti, L.; Schuchman, E.H.; Furlan, R.; Clementi, E.; et al. Acid Sphingomyelinase Activity Triggers Microparticle Release from Glial Cells. EMBO J. 2009, 28, 1043–1054.
- Charras, G.; Paluch, E. Blebs Lead the Way: How to Migrate without Lamellipodia. Nat. Rev. Mol. Cell Biol. 2008, 9, 730–736.
- Babiychuk, E.B.; Monastyrskaya, K.; Potez, S.; Draeger, A. Blebbing Confers Resistance against Cell Lysis. Cell Death Differ. 2011, 18, 80–89.
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379.
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and INFLAMMATION. Immunity 2017, 47, 15–31.
- Inoue, K. Microglial Activation by Purines and Pyrimidines. Glia 2002, 40, 156–163.
- Shieh, C.; Heinrich, A.; Serchov, T.; van Calker, D.; Biber, K. P2X7-Dependent, but Differentially Regulated Release of IL-6, CCL2, and TNF-α in Cultured Mouse Microglia. Glia 2014, 62, 592–607.
- Savio, L.E.B.; de Andrade Mello, P.; Da Silva, C.G.; Coutinho-Silva, R. The P2X7 Receptor in Inflammatory Diseases: Angel or Demon? Front. Pharmacol. 2018, 9, 52.
- Sanz, J.M.; Di Virgilio, F. Kinetics and Mechanism of ATP-Dependent IL-1β Release from Microglial Cells. J. Immunol. 2000, 164, 4893–4898.
- Suzuki, T.; Hide, I.; Ido, K.; Kohsaka, S.; Inoue, K.; Nakata, Y. Production and Release of Neuroprotective Tumor Necrosis Factor by P2X7 Receptor-Activated Microglia. J. Neurosci. 2004, 24, 1–7.
- Facci, L.; Barbierato, M.; Zusso, M.; Skaper, S.D.; Giusti, P. Serum Amyloid A Primes Microglia for ATP-Dependent Interleukin-1β Release. J. Neuroinflamm. 2018, 15, 164.
- Tewari, M.; Khan, M.; Verma, M.; Coppens, J.; Kemp, J.M.; Bucholz, R.; Mercier, P.; Egan, T.M. Physiology of Cultured Human Microglia Maintained in a Defined Culture Medium. ImmunoHorizons 2021, 5, 257–272.
- Ferrari, D.; Pizzirani, C.; Adinolfi, E.; Lemoli, R.M.; Curti, A.; Idzko, M.; Panther, E.; Di Virgilio, F. The P2X7 Receptor: A Key Player in IL-1 Processing and Release. J. Immunol. 2006, 176, 3877–3883.
- Munoz, F.M.; Patel, P.A.; Gao, X.; Mei, Y.; Xia, J.; Gilels, S.; Hu, H. Reactive Oxygen Species Play a Role in P2X7 Receptor-Mediated IL-6 Production in Spinal Astrocytes. Purinergic Signal. 2020, 16, 97–107.
- Kim, S.Y.; Moon, J.H.; Lee, H.G.; Kim, S.U.; Lee, Y.B. ATP Released from β-Amyloid-Stimulated Microglia Induces Reactive Oxygen Species Production in an Autocrine Fashion. Exp. Mol. Med. 2007, 39, 820–827.
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437.
- Adinolfi, E.; De Marchi, E.; Orioli, E.; Pegoraro, A.; Di Virgilio, F. Role of the P2X7 Receptor in Tumor-Associated Inflammation. Curr. Opin. Pharmacol. 2019, 47, 59–64.
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899.
- Gilbert, S.; Oliphant, C.; Hassan, S.; Peille, A.; Bronsert, P.; Falzoni, S.; Di Virgilio, F.; McNulty, S.; Lara, R. ATP in the Tumour Microenvironment Drives Expression of nfP2X7, a Key Mediator of Cancer Cell Survival. Oncogene 2019, 38, 194–208.
- Amoroso, F.; Capece, M.; Rotondo, A.; Cangelosi, D.; Ferracin, M.; Franceschini, A.; Raffaghello, L.; Pistoia, V.; Varesio, L.; Adinolfi, E. The P2X7 Receptor is a Key Modulator of the PI3K/GSK3β/VEGF Signaling Network: Evidence in Experimental Neuroblastoma. Oncogene 2015, 34, 5240–5251.
- Di Virgilio, F.; Adinolfi, E. Extracellular Purines, Purinergic Receptors and Tumor Growth. Oncogene 2017, 36, 293–303.
- Scarpellino, G.; Genova, T.; Munaron, L. Purinergic P2X7 Receptor: A Cation Channel Sensitive to Tumor Microenvironment. Recent Patents Anti-Cancer Drug Discov. 2019, 14, 32–38.
- Arnaud-Sampaio, V.F.; Rabelo, I.L.A.; Ulrich, H.; Lameu, C. The P2X7 Receptor in the Maintenance of Cancer Stem Cells, Chemoresistance and Metastasis. Stem Cell Rev. Rep. 2019, 16, 288–300.
- Hui, L.; Chen, Y. Tumor Microenvironment: Sanctuary of the Devil. Cancer Lett. 2015, 368, 7–13.
- Bianchi, G.; Vuerich, M.; Pellegatti, P.; Marimpietri, D.; Emionite, L.; Marigo, I.; Bronte, V.; Di Virgilio, F.; Pistoia, V.; Raffaghello, L. ATP/P2X7 Axis Modulates Myeloid-Derived Suppressor Cell Functions in Neuroblastoma Microenvironment. Cell Death Dis. 2014, 5, e1135.
- Adinolfi, E.; Raffaghello, L.; Giuliani, A.L.; Cavazzini, L.; Capece, M.; Chiozzi, P.; Bianchi, G.; Kroemer, G.; Pistoia, V.; Di Virgilio, F. Expression of P2X7 Receptor Increases In Vivo Tumor Growth. Cancer Res 2012, 72, 2957–2969.
- Bergamin, L.S.; Capece, M.; Salaro, E.; Sarti, A.C.; Falzoni, S.; Pereira, M.S.L.; De Bastiani, M.A.; Scholl, J.N.; Battastini, A.M.O.; Di Virgilio, F. Role of the P2X7 Receptor in In Vitro and In Vivo Glioma Tumor Growth. Oncotarget 2019, 10, 4840–4856.
- Virgilio, F.D. Purines, Purinergic Receptors, and Cancer. Cancer Res. 2012, 72, 5441–5447.
- Zhang, Y.; Li, F.; Wang, L.; Lou, Y. A438079 Affects Colorectal Cancer Cell Proliferation, migration, apoptosis, and pyroptosis by inhibiting the P2X7 receptor. Biochem. Biophys. Res. Commun. 2021, 558, 147–153.
- Kan, L.K.; Williams, D.; Drummond, K.; O’Brien, T.; Monif, M. The Role of Microglia and P2X7 Receptors in Gliomas. J. Neuroimmunol. 2019, 332, 138–146.
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in Dendritic Cells Induces IL-1β–Dependent Adaptive Immunity against Tumors. Nat. Med. 2009, 15, 1170–1178.
- Adinolfi, E.; Capece, M.; Franceschini, A.; Falzoni, S.; Giuliani, A.L.; Rotondo, A.; Sarti, A.C.; Bonora, M.; Syberg, S.; Corigliano, D.; et al. Accelerated Tumor Progression in Mice Lacking the ATP Receptor P2X7. Cancer Res 2015, 75, 635–644.
- McLarnon, J.G. Roles of Purinergic P2X7 Receptor in Glioma and Microglia in Brain Tumors. Cancer Lett. 2017, 402, 93–99.
- Hoelzinger, D.B.; Demuth, T.; Berens, M.E. Autocrine Factors That Sustain Glioma Invasion and Paracrine Biology in the Brain Microenvironment. JNCI J. Natl. Cancer Inst. 2007, 99, 1583–1593.
- Cekic, C.; Day, Y.-J.; Sag, D.; Linden, J. Myeloid Expression of Adenosine A2A Receptor Suppresses T and NK Cell Responses in the Solid Tumor Microenvironment. Cancer Res 2014, 74, 7250–7259.
- Tarassishin, L.; Lim, J.; Weatherly, D.B.; Angeletti, R.H.; Lee, S.C. Interleukin-1-Induced Changes in the Glioblastoma Secretome Suggest Its Role in Tumor Progression. J. Proteom. 2014, 99, 152–168.
- Illes, P. P2X7 Receptors Amplify CNS Damage in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 5996.
- Tewari, M.; Seth, P. Emerging Role of P2X7 Receptors in CNS Health and Disease. Ageing Res. Rev. 2015, 24, 328–342.
- Hogquist, K.A.; Nett, M.A.; Unanue, E.R.; Chaplin, D.D. Interleukin 1 is Processed and Released during Apoptosis. Proc. Natl. Acad. Sci. USA 1991, 88, 8485–8489.
- Brough, D.; Le Feuvre, R.A.; Iwakura, Y.; Rothwell, N.J. Purinergic (P2X7) Receptor Activation of Microglia Induces Cell Death via an Interleukin-1-Independent Mechanism. Mol. Cell. Neurosci. 2002, 19, 272–280.
- Mehta, V.B.; Hart, J.; Wewers, M.D. ATP-stimulated Release of Interleukin (IL)-1β and IL-18 Requires Priming by Lipopolysaccharide and Is Independent of Caspase-1 Cleavage. J. Biol. Chem. 2001, 276, 3820–3826.
- Denlinger, L.C.; Fisette, P.L.; Sommer, J.A.; Watters, J.J.; Prabhu, U.; Dubyak, G.R.; Proctor, R.A.; Bertics, P.J. Cutting Edge: The Nucleotide Receptor P2X7 Contains Multiple Protein- and Lipid-Interaction Motifs Including a Potential Binding Site for Bacterial Lipopolysaccharide. J. Immunol. 2001, 167, 1871–1876.
- Bartlett, R.; Yerbury, J.J.; Sluyter, R. P2X7 Receptor Activation Induces Reactive Oxygen Species Formation and Cell Death in Murine EOC13 Microglia. Mediat. Inflamm. 2013, 2013, 271813.
- Loureiro, A.V.; Moura-Neto, L.I.; Martins, C.S.; Silva, P.I.M.; Lopes, M.B.; Leitão, R.F.C.; Coelho-Aguiar, J.M.; Moura-Neto, V.; Warren, C.A.; Costa, D.V.; et al. Role of Pannexin-1-P2X7R Signaling on Cell Death and Pro-Inflammatory Mediator Expression Induced by Clostridioides Difficile Toxins in Enteric Glia. Front. Immunol. 2022, 13, 956340.
- Cavaliere, F.; Dinkel, K.; Reymann, K. Microglia Response and P2 Receptor Participation in Oxygen/Glucose Deprivation-Induced Cortical Damage. Neuroscience 2005, 136, 615–623.
- Cavaliere, F.; D’Ambrosi, N.; Sancesario, G.; Bernardi, G.; Volonté, C. Hypoglycaemia-Induced Cell Death: Features of Neuroprotection by the P2 Receptor Antagonist Basilen Blue. Neurochem. Int. 2000, 38, 199–207.
- Eyo, U.B.; Miner, S.A.; Ahlers, K.E.; Wu, L.-J.; Dailey, M.E. P2X7 Receptor Activation Regulates Microglial Cell Death during Oxygen-Glucose Deprivation. Neuropharmacology 2013, 73, 311–319.
- Jia, X.; Xie, L.; Liu, Y.; Liu, T.; Yang, P.; Hu, J.; Peng, Z.; Luo, K.; Du, M.; Chen, C. Astragalus polysaccharide (APS) Exerts Protective Effect against Acute Ischemic Stroke (AIS) through Enhancing M2 Micoglia Polarization by Regulating Adenosine Triphosphate (ATP)/Purinergic Receptor (P2X7R) Axis. Bioengineered 2022, 13, 4468–4480.
More