Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Cornelius Fernandez James and Version 2 by Mona Zou.
Metabolic dysfunction-associated fatty liver disease (MAFLD) has now affected nearly one-third of the global population and has become the number one cause of chronic liver disease in the world because of the obesity pandemic. Chronic hepatitis resulting from hepatitis B virus (HBV) and hepatitis C virus (HCV) remain significant challenges to liver health even in the 21st century. The co-existence of MAFLD and chronic viral hepatitis can markedly alter the disease course of individual diseases and can complicate the management of each of these disorders. A thorough understanding of the pathobiological interactions between MAFLD and these two chronic viral infections is crucial for appropriately managing these patients.
- metabolic dysfunction-associated fatty liver disease (MAFLD)
- chronic viral hepatitis
- hepatitis B virus (HBV)
- hepatitis C virus (HCV)
- hepatic fibrosis
- cirrhosis
- hepatocellular carcinoma
1. MAFLD and Chronic HBV Infection
Epidemiology
According to a World Health Organization (WHO) report, in 2015, more than 250 million people globally were suffering from chronic hepatitis B (CHB) infection [1][7]. Additionally, 887,000 people died from complications related to CHB, including cirrhosis and liver cancer, in the same year. These data underscores the immense burden that CHB places on global public health. There is no direct evidence that CHB is associated with an increased risk of hepatic steatosis. Several meta-analyses have examined this phenomenon. In a meta-analysis of 17 studies, which included 4100 HBV-infected patients and 8 of which also included 945 HCV-infected patients, it was reported that approximately 29.6% of patients with HBV developed fatty liver, like in the general population [2][8]. The same study observed that 60% of the patients with HCV developed fatty liver. Moreover, the study observed a statistically significant positive association with the male sex (OR 1.74, 95% CI [1.28–2.38], p < 0.001) and body mass index (SMD 2.17, 95% CI [1.23, 3.11], p < 0.001); and a negative association with HBV-DNA (SMD −74.12, 95% CI [−82.93, −65.31], p < 0.001). This strong negative association between HBV-DNA and steatosis may indicate a protective effect of HBV infection on steatosis. Another meta-analysis of 54 studies, involving 28,648 CHB patients, found a pooled prevalence of hepatic steatosis of up to 32.8% [3][9]. A more recent meta-analysis, which included 98 studies and 48,472 patients, demonstrated an even higher global prevalence of hepatic steatosis among CHB patients, reaching 34.93% [4][10].
2. Effect of MAFLD on CHB Infection and Chronic Liver Disease Progression
MAFLD is associated with increased Th17 cell-related gene expression, increased IL-21 levels, activation of T and B cells, production of inflammatory cytokines, elimination of HBV proliferation with resultant immune clearance of HBV DNA, and HbeAg [5][11]. The NASH stage of MAFLD is associated with increased expression of toll-like receptors (TLRs) in hepatocytes, Kupffer cells (KCs), hepatic stellate cells (HSCs), sinusoidal endothelial cells, and hepatic dendritic cells (DCs) [6][12]. Lipopolysaccharide (LPA) induces activation of the TLR4 and myeloid differentiation factor 88 (MyD88)-mediated pathways in obese individuals [7][13]. Activation of the TLR4/Myd88 pathway contributes to the activation of HSCs and the production of chemokines, which recruits further KCs [8][14]. TLR4 activation in KCs induces the secretion of pro-inflammatory cytokines (IL-1, IL-6, IL-8, TNF-α, and chemokines) and profibrogenic factors (TGF-β) to activate the inflammation–fibrosis–carcinoma sequence [8][14]. TLR4/MyD88 signaling also induces the production of IFN-β, IL-6, and TNF-α to inhibit HBV replication [7][13]. Thus, activation of innate immunity through TLR signaling is associated with the inhibition of HBV replication and the retardation of the progression of MAFLD to NASH, fibrosis, and HCC [9][15].
MAFLD-associated metabolic stress could reduce peroxisome proliferator-activated receptor–gamma coactivator 1 alpha (PGC-1α), which in turn could inhibit HBV replication and induce Fas-mediated apoptosis of HBV-infected cells, resulting in HBV-clearance and reduction of HBV-related liver disease progression [10][16]. CHB is associated with a decreased risk of hyperlipidemia [11][12][17,18] and raised serum adiponectin levels [13][19], which could contribute to a lower risk of hepatic steatosis.
On the other hand, the production of saturated fatty acid–palmitic acid as a metabolic component of MAFLD could be associated with impaired function of hepatic DCs and impaired HBsAg processing/presentation, leading to inadequate immune response/HBV-clearance and subsequent development of severe HBV-related liver disease progression [14][20].
3. Effects of CHB Infection on the MAFLD and Chronic Liver Disease Progression
Some of the transcription factors (including CEBP [15][22], CREB [16][23], HNF3 [17][24], HNF4 [18][25], FXR [19][26], RXR [20][27], and PPAR [21][28]) involved in the transcription of HBV DNA are involved in hepatic glucose, lipid, bile acid, and xenobiotic metabolism [21][28] may either inhibit or induce regeneration, inflammation, fibrosis, and malignant transformation of hepatic cells. Differential expressions of IL-13, G-CSF, CCL11, IL-6, and IL-4 are thought to play a role in developing steatosis and fibrosis in patients with CHB infection. IL-13 facilitates hepatic steatosis and fibrosis, the latter through mechanisms including the stimulation of TGF-β1 gene expression [22][29] and through activation of the JAK-STAT-6 pathway, in turn results in the production of CCL11, an eosinophil chemotactic protein [23][30]. CCL11-mediated hepatic eosinophilic infiltration and activation results in hepatic steatosis and fibrosis [24][31]. G-CSF ameliorates hepatic steatosis by reducing the expression of SREBP-1c [25][32]. IL-4 and IL-6 protect against hepatic fibrosis [26][33], IL-4 through secretion of matrix metalloproteinase-12 (MMP-12) [27][34], and IL-6 through the promotion of proliferation/survival of HSCs [28][35].
In patients with CHB infection, hepatitis B protein X (HBx)—a 17 kDa soluble protein coded by the HBV DNA induces expression of various genes related to lipid accumulation including PPAR [29][36], SREBP [29][36], FABP1 [30][37], LXR [31][38], and FATP2 [32][39], thereby promoting lipogenesis. HBx also stimulates various transcription factors, including STAT3, NF-κβ, PI3K/AKT, and Src [33][40], which promote hepatocyte proliferation [33][40], inhibit apoptosis [33][40], and stimulate inflammation [34][41], thus leading to the development of HCC. Moreover, the pre-S1 domain of the HBV envelope binds to sodium taurocholate cotransporting polypeptide (NCTP), limiting the function of NCTP, thus promoting compensatory bile acid synthesis, cholesterol provision, and hepatic steatosis [35][42]. Steatosis associated with MAFLD, and the resultant oxidative stress might generate an intra-hepatic pro-fibrotic and pro-cancerous environment [36][43]. Additionally, CHB-associated deficiency of PML (promyelocytic leukemia protein) results in altered lipid metabolism and steatosis-associated carcinogenesis [37][44]. Reduced levels of global DNA methylation in patients with concurrent MAFLD and CHB lead to chromosomal abnormality, instability, fragility, and HCC development [38][45].
Hepatic steatosis was observed in nearly 18% of patients with biopsy-proven CHB infection [39][46]. Steatosis had an independent association with body mass index and fasting blood glucose levels, and it does not correlate with the degree of hepatic fibrosis [39][46]. There is a possible genetic susceptibility to develop steatosis in CHB infection, with the rs1010023 polymorphism in the PNPLA3 gene and rs58542926 polymorphism in the TM6SF2 gene increasing the tendency to develop MAFLD among patients with CHB infection [36][43]. HBx could play an important role in increasing the risk of HBV-induced steatosis. On the other hand, the reduced risk of hyperlipidemia and the increased adiponectin levels could reduce the risk of HBV-induced steatosis. Although MAFLD is associated with lower HBV viral load and with an increased rate of HBsAg clearance, both CHB and MAFLD could act synergistically to promote the progression of liver disease, causing hepatocyte injury, inflammation, fibrosis, and HCC. Figure 1 shows the pathobiological interlink between chronic HBV infection and metabolic dysfunction and the impact of MAFLD on HBV replication.
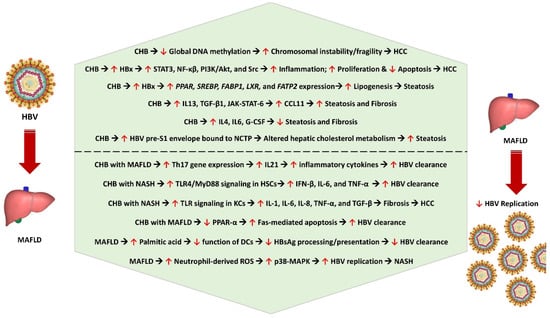
Figure 1. Pathobiological interlink between CHB and metabolic dysfunction and the impact of MAFLD on HBV replication. CHB—chronic hepatitis B, HCC—hepatocellular carcinoma, HBx—hepatitis B protein X, STAT3—signal transducer and activator of transcription 3, NF-kβ—nuclear factor kappa B subunit, PI3K/AKT—phosphoinositide 3-kinase/protein kinase B, PPAR—peroxisome proliferator-activated receptor gene, SREBP—sterol regulatory element-binding protein gene, FABP1—fatty acid-binding protein 1 gene, LXR—liver X receptor gene, FATP2—fatty acid transport protein 2 gene, IL13—interleukin 13, TGF-β1—transforming growth factor beta 1, JAK-STAT-6—Janus kinase-signal transducer and activator of transcription 6, CCL11—C-C motif ligand 11 (eosinophil chemotactic protein or eotaxin-1), IL4—interleukin 4, IL6—interleukin 6, G-CSF—granulocyte colony-stimulating factor, NCTP—sodium taurocholate cotransporting polypeptide, Th17—T helper 17 cell, IL21—interleukin 21, TLR4/Myd88—Toll-like receptor-myeloid differentiation factor 88, IFN-β—interferon beta, KCs—Kupffer cells, HSCs—hepatic stellate cells, IL8—interleukin 8, TNF-α—tumor necrosis factor alpha, Fas or FasR—Fas receptor (apoptosis antigen 1), DCs—dendritic cells, HbsAg—hepatitis B surface antigen, ROS—reactive oxygen species, p38-MAPK—p38-mitogen-activated protein kinase, NASH—nonalcoholic steatohepatitis.
A retrospective study involving 1076 CHB patients with a median follow-up period of 9.8 years evaluated the importance of MAFLD in patients with CHB [40][47]. The study observed that MAFLD is associated with reduced event-free (aHR 2.00, 95% CI 1.26–3.19), HCC-free (aHR 1.93, 95% CI 1.17–3.21), and transplant-free survival (aHR 1.80, 95% CI 0.98–3.29), implying higher risk for liver-related events and death. A prospective study of 10,546 CHB patients observed that after a median follow-up period of 5.1 years, MAFLD is associated with a 58% reduced risk of HCC (adjusted hazard ratio or aHR 0.42, 95% CI 0.25–0.68, p < 0.001) [41][48]. The steatosis and metabolic dysfunction had distinctive effects on the risk for HCC. While steatosis was protective against HCC (aHR 0.45, 95% CI 0.30–0.67, p < 0.001), a greater burden of metabolic dysfunction increased the HCC risk (aHR 1.40 per dysfunction increase, 95% CI 1.19–1.66, p < 0.001) [41][48].
Management
The management of MAFLD in patients with CHB involves a multifaceted approach. Traditional liver biopsy, considered the gold standard for diagnosis of hepatic steatosis, is associated with a high risk of internal bleeding [42][55], making non–invasive methods a more appropriate approach. One such method is the controlled attenuation parameter (CAP) via fibro-scan [43][44][56,57], which measures attenuation during ultrasonography to estimate the degree of steatosis. CAP has a relatively low cost and is suitable for most first-line clinical settings [45][58]. In CHB, patients’ CAP demonstrated a high degree of accuracy for steatosis assessment compared to other noninvasive methods [46][47][59,60]. It has been used in predicting the presence and severity of MAFLD in CHB patients [48][61].
CHB management requires antiviral treatments such as nucleotide analogs like tenofovir alafenamide or entecavir to suppress viral replication [49][62], although a cure is often difficult. Patients with concurrent MAFLD may experience variations in viral activity and liver enzymes due to the presence of NASH [50][63]. Conflicting evidence exists in the response to treatment in patients with co-existent MAFLD and CHB. While some studies indicate lower treatment response in CHB patients with hepatic steatosis, others show comparable responses. Monitoring serum ALT and HBV DNA levels and timely intervention for poor responders are crucial for managing CHB in the presence of MAFLD [51][52][64,65].
Acute intervention for concurrent MAFLD is crucial, given its adverse impact on overall health. Lifestyle modifications, including strict diet control aiming at weight loss and adherence to certain dietary practices, such as a hypocaloric diet and avoidance of food high in saturated fats or ultra-processed foods, coupled with regular exercise, form the cornerstones of therapy [53][54][66,67]. Several pharmacological treatment options for steatohepatitis are currently being developed, such as semaglutide [55][68], lanifibranor (pan-peroxisome proliferator-activated receptor agonist) [56][69], resmetirom (selective thyroid hormone receptor-β agonist) [57][58][70,71] and obeticholic acid (selective farnesoid X receptor agonist) [59][60][72,73], with some promising results, but their routine use in CHB patients with concurrent MAFLD requires further evaluation.
Improvement of hepatic steatosis may affect HBV replication, necessitating careful monitoring during metabolic correction. Factors like diabetes mellitus, obesity, and dyslipidemia contribute to the progression of both MAFLD and CHB infection [61][74], making the aggressive management of both conditions essential. These metabolic risk factors are independently associated with liver disease progression, hepatocarcinogenesis, and overall mortality in CHB patients [62][63][75,76]. Therefore, addressing metabolic dysfunction is the key to improving co-existent CHB in patients with MAFLD.
4. MAFLD and Chronic HCV Infection
Epidemiology
According to global estimates, approximately 71.1 million people have chronic hepatitis C virus infection, with a global prevalence of 1% in 2015 [64][77]. Globally, the most common HCV genotype is genotype 1 (nearly 50% of all adults with HCV infection), followed by genotypes 3, 2, 4, 6, and 5 respectively [65][78]. HCV infection, especially genotype 3, is well known to be associated with hepatic steatosis. Genotype 3 is highly steatogenic [66][79], and it exhibits a steatosis prevalence of up to 86% while other phenotypes possess a steatosis prevalence of around 50% [67][80]. The mean prevalence of steatosis in chronic HCV is around 55% across all HCV genotypes [67][80]. HCV genotype 3 is reported to exert a direct cytopathic effect on the liver in direct proportion to the viral load, even in the absence of other metabolic risk factors like visceral obesity and/or diabetes mellitus [1][7]. The term ‘viral steatosis’ is used for this entity [67][80].
With the change in nomenclature from NAFLD to MAFLD, those patients with HCV infection who also meet the criteria for the diagnosis of MAFLD are classified as hepatitis C with MAFLD. Thus, there are now two types of HCV: hepatitis C with MAFLD and hepatitis C without MAFLD. The term ‘metabolic steatosis’ is used for the entity seen in patients with hepatitis C and MAFLD [67][80]. Contrary to metabolic steatosis, ‘viral steatosis’ is associated with reduced LDL cholesterol and triglyceride levels [68][81]. Genotypes 1, 2, and 4 essentially promote insulin resistance associated with host metabolic risk factors, including visceral obesity [66][79]. MAFLD patients with hepatitis C have a higher risk for advanced hepatic fibrosis but with a similar atherosclerotic CVD risk in comparison to those with MAFLD alone without CHC infection (CHC) [69][82].
A recent Australian study [70][83] observed a 43.1% prevalence of MAFLD in patients with CHC infection in contrast to the global prevalence of MAFLD of 25% in the general population [71][84]. This dual etiology group is associated with an increased risk for hepatic injury, inflammation, and fibrosis (all p < 0.001). This study observed that those with CHC and lean MAFLD had a similar rate of advanced fibrosis (31.6%) in comparison to those who had obesity and/or diabetes mellitus (31.8% and 46.2%, respectively, with p = 0.325). However, those with dual etiology are at a greater risk of developing advanced fibrosis and HCC even after HCV clearance, implying that managing MAFLD is equally as important as HCV clearance to prevent the progression of hepatic disease and death from HCC or cardiovascular disease [71][84].
5. Effect of MAFLD on CHC Infection and Chronic Liver Disease Progression
Lipid droplets are involved in the replication and virion assembly of HCV, and stimulation of de novo lipogenesis (DNL) via MAFLD (and CHC) facilitates the entry of the virus into the hepatocytes [72][86]. Moreover, upon release from hepatocytes, the mature HCVs in circulation are complexed with lipoproteins [73][87]. A complex metabolic network exists in the fatty liver to regulate HCV replication. While saturated and monounsaturated fatty acids are required for replication, polyunsaturated fatty acids inhibit HCV RNA replication [74][88]. Lipid peroxidation, a feature of NASH, inhibits HCV replication [75][89]. HCV-infected cells have phosphatidylcholines and triglycerides with longer fatty acyl chains [76][90]. Knocking down fatty acid elongases [76][90], fatty acid desaturases [76][90], or phosphatidyl ethanolamine transferase [77][91] (PEMT) can inhibit HCV RNA replication.