Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Diana Camelia Nuta and Version 2 by Jason Zhu.
Microorganisms participating in the development of biofilms exhibit heightened resistance to antibiotic treatment, therefore infections involving biofilms have become a problem in recent years as they are more difficult to treat. Consequently, research efforts are directed towards identifying novel molecules that not only possess antimicrobial properties but also demonstrate efficacy against biofilms. While numerous investigations have focused on antimicrobial capabilities of Schiff bases, their potential as antibiofilm agents remains largely unexplored.
- Schiff base
- antibiofilm
- antimicrobial
- imines
1. Introduction
Clinically relevant microbial biofilms are defined as “aggregated microbial cells surrounded by a polymeric self-produced matrix, which may contain host components”, suspended or attached to a surface [1]. Biofilm-related infections attracted the attention of scientists 50 years ago in the context of cystic fibrosis, and their impact on the medical field has grown ever since [2]. These infections may be tissue-related (chronic otitis media, chronic sinusitis, chronic laryngitis, dental plaque, endocarditis, cystic fibrosis, kidney stones, biliary tract infections, urinary tract infections, osteomyelitis, wound infections, etc.) or associated with medical devices (contact lenses, endotracheal tubes, cardiac devices or catheters) [1][3][1,3]. Some examples of biofilm forming pathogens are: Pseudomonas aeruginosa, Staphylococcus aureus, Haemophilus influenzae, Staphylococcus epidermidis, Streptococci, Enterococci and Candida spp. [4].
Biofilm formation requires four stages: (i) attachment of the mobile microorganism to a surface, (ii) colonization, (iii) development and maturation of biofilm and (iv) dispersion and propagation [5][6][5,6]. Attachment is mediated by cilli, flagella, surface proteins of microorganisms and rugosity of the surface [7]. It is reversible at first and then becomes irreversible, triggering transcription of specific genes for signalling molecules and extracellular polymeric substances (EPS). Colonization involves growth and division processes and EPS synthesis [5]. A mature biofilm consists of three layers: the biofilm nucleus, membranes of basal microorganisms and external mobile planktonic cells. It is a complex mixture of water, microbial cells, proteins, aminoacids and polysaccharides [6]. Dispersion is mediated by external factors or by self-digestion and contributes to dissemination of infection [7]. There are two important features of biofilm (sessile) growth compared with the free-floating (planktonic) state that contributes to pathogenicity: increased tolerance to antibiotic treatment and persistence in the host, despite inflammation and immune response [1]. The major consequence is that biofilm infections are hard to treat and usually become chronic [8].
Antibiotherapy is active on planktonic microbial cells, but its effectiveness against sessile states is variable, as established biofilms are usually recalcitrant to conventional antibiotics [9]. Treatment may require higher doses of antimicrobials, prolonged duration [8][10][8,10], combination therapy [11][12][11,12] or special modes of administration (nebulized antibiotics) [13].
There is a constant need to develop alternative antibiofilm strategies, and extensive research has been conducted in this direction [10]. Antibiofilm small molecules are relevant because they target stages in biofilm development which are different to those of normal planktonic state [9]. The mechanisms (Figure 1) may involve: blocking microbial adhesion (biocides [14], antibiotics [15] and impregnated coatings [16]), inhibition of microbial communication (quorum sensing inhibitors and quorum quenching) [17] and killing cells inside the biofilm (persisters or non-growing cells) (cisplatin, cis-2-decenoic acid, colistin, mytomicin C) [18][19][20][21][18,19,20,21]. In particular, strategies like quorum sensing inhibition may prove useful because they do not necessarily affect bacterial growth but they reduce virulence, thus increasing susceptibility of microorganisms to antibiotics and to host immune cells without the risk of antibiotic resistant [17].
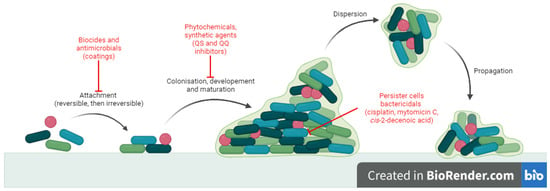
Figure 1.
Antibiofilm mechanisms of action for small molecules.
Schiff bases are compounds with the structure R’N=CR2 (R’ ≠ H) [22], traditionally formed in the reaction of alkyl/aryl aldehydes or ketones with primary amines [23]. Many considered them to be synonymous with azomethines (RN=CR2, R ≠ H), both being a particular case of imines (RN=CR2, R = H, hydrocarbonyl) [22]. They are all part of carbonyl compound derivatives, formed in the reaction with basic nucleophiles (amines and their derivatives—hydroxylamine, hydrazine, N-acyl-hydrazine and semicarbazide). Therefore, this class of compounds also includes oximes (RR’C=NOH), hydrazones (R1R2C=N-NH2), N-acyl-hydrazones (R1R2C=N-NH-CO-R) and semicarbazones (R1R2C=NNH-(CO)-NH2) [14].
Schiff bases have numerous applications including coordination chemistry [23], catalysis [24], chemosensors [25] and intermediates in synthesis [26][27][26,27]. They also exhibit a variety of biological applications: antibacterial [28][29][30][31][32][33][34][28,29,30,31,32,33,34], antifungal [34][35][36][34,35,36], antiviral [37][38][37,38], antimalarial [39][40][39,40], antituberculosis [41][42][43][41,42,43], anthelmintic [44][45][44,45], urease inhibitors [46][47][48][46,47,48], anticancer [49][50][51][49,50,51], antidyslipidemic [52], antidiabetic [53][54][53,54], antidepressant [55][56][57][55,56,57], anticonvulsant [58][59][58,59], neurodegenerative disorder treatment [60][61][60,61], anti-inflammatory [62] and antioxidant [53][54][53,54].
Human and veterinary therapy benefits from several antibacterial drugs recognized as Schiff bases. In multidrug-resistant tuberculosis (MDR-TB), a longer treatment regimen includes two Schiff bases which act on Mycobacterium tuberculosis cell wall: bacteriostatic terizidone (Figure 2), a cycloserine derivative, analogue of D-alanine and anti-leprosy clofazimine, which is an iminophenazine [63][64][63,64].
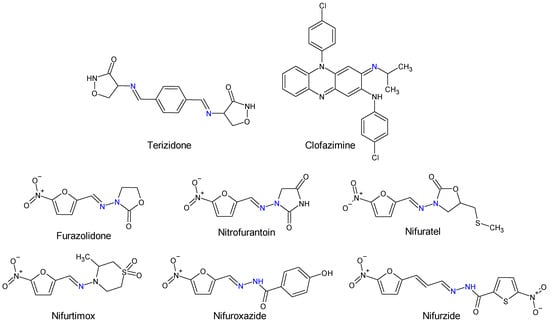
Figure 2.
Structure of Schiff base and
N
-acyl-hydrazone medicines.
Oximes and hydrazones are moieties frequently used in medicinal chemistry. Examples of oxime drugs utilized in antimicrobial therapy include: cephalosporins (second-generation—cefuroxime, third-generation—cefdinir, cefixime, cefpodoxime, ceftazidime, cefmenoxime, ceftizoxime, ceftriaxone, cefotaxime, cefpirome, fourth-generation—cefepime, fifth-generation—ceftaroline, cefiderocol) [65][66][67][68][65,66,67,68], as well as antifungal (oxiconazole) [69], antiviral (enviroxime, zinviroxime) [70][71][70,71] and anti-infective (nifuroxime) medications [72] (Figure 2 and Figure 3).
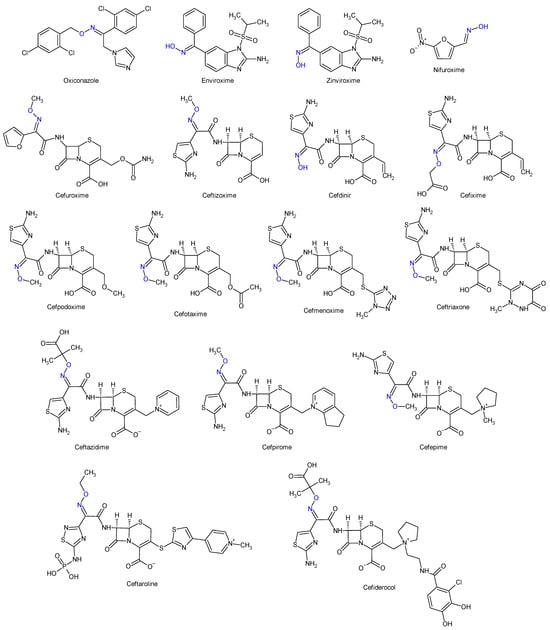
Figure 3.
Structure of oxime medicines.
N-acyl-hydrazones derivatives of 5-nitrofuran are prodrugs which act against different types of pathogens [73]. Some examples are nifuratel (antibacterial, antifungal, antitrichomonal agent) [74], furazolidone (antiprotozoal agent, gynaecological antiinfective and antiseptic) [75][76][75,76], nifurzide, nifuroxazide (intestinal antiinfectives, antidiarrheal agents) [77], nitrofurantoin (antibacterial) [78] and nifurtimox (antitrypanosomal and antileshmanial agent) [79]. Along with their stated antiinfective indications, studies have explored other possible applications of these drugs. Nifuratel—activity against Leishmania spp. [80][81][80,81], nifuroxazide—quorum sensing and biofilm inhibition [82], antischistosomal activity [83]. N-acyl-hydrazones 5-nitrofurans also exhibit anticancer properties, inhibiting different pathways in cancer cell cycles: signal transducer and activator of transcription 3 (STAT3) (nifuroxazide [84], nifuratel [85]), aldehyde dehydrogenase 1 (ALDH1) (nifuroxazide [86]) and nuclear factor kappa B signalling (furazolidone [87]) (Figure 2 and Figure 3).
2. A Possible Strategy to Combat Biofilm-Related Infections
Due to their ease of synthesis and their wide range of applications, salicylaldehyde Schiff bases are frequently cited in the relevant literature. These compounds demonstrate antimicrobial potential, both as simple ligands and as metal complexes [56][57][58][56,57,58]. The antimicrobial activity is directly influenced by substitutions on the salicyl moiety, with halogenation exerting a noticeable impact in particular [58]. Taurine-5-bromosalicylaldehyde Schiff base (TBSSB) is a potassium salt of 2-{[1-(5-bromo-2-hydroxyphenyl)-meth-(Z)-ylidene]-amino} ethanesulfonic acid, with antistaphylococcal [88][97] and antimycobacterial potential [89][98] that is active against both planktonic and sessile forms. TBSSB was bactericidal against S. aureus (MIC 32 μg/mL), affecting membrane integrity and also preventing biofilm formation at 8 μg/mL [88][97]. The sulfonic acid group seems essential to antistaphylococcal activity [88][97]. The antimycobacterial effect was even better. TBSSB completely inhibited M. smegmatis mc2155 growth at > 60 μg/mL, presenting greater cell wall destruction compared with S. aureus alongside alterations in cell division. Additionally, it exhibited dose-dependent inhibition of Mycobacterium biofilm formation [31]. p-Aminobenzoic acid (PABA) is an amino acid derivative, implicated in folate biosynthesis in microbial cells [90][133]. Due to its importance for bacterial viability, it serves as a target for antimicrobial therapy [91][134]. Therefore, obtaining hybrid molecules is a direction of molecular development [92][135] in the search for new anti-infective agents. Starting from a series of Schiff base derivatives of p-aminobenzoic acid and halogenated salicylaldehydes (compound 1), me-too analogues were synthesized and tested for antimicrobial, antibiofilm and cytotoxicity activities [93][99]. The design approaches were as follows: isomerization (m-aminobenzoic acid (MABA) derivatives 2) esterification (methyl esters 3, and ethyl esters 4) amide formation (N-phenylamides 5) duplication of azomethine bond (3,5-diaminobenzoic acid (DABA) derivatives 6). The Schiff bases obtained were active against Gram-positive strains, having MIC from 7.81 μM. The corresponding amines presented no antimicrobial effect. Diiodo derivatives (2b, 3b and 6b) were comparable in action to bacitracin (SA: MIC 7.81 μM, EF: 15.62 μM). No activity was observed against Mycobacterium strains. Regarding antifungal activity, the analogues surpassed the original PABA Schiff bases. Derivatives 2, 5 and 6 exhibited broad-spectrum activity, C. albicans and T. interdigitale being the most susceptible. The best results were obtained for diiodo analogues (2b, 5b, 6b), having MICs comparable to fluconazole (CA: 6.5 μM). The antibiofilm evaluation was performed on two strong biofilm producers: methicillin-resistant S. aureus ATCC 43300 and S. epidermidis ATCC 1228. Compound 3b (methyl (E)-4-[(2-hydroxy-3,5-diiodobenzylidene)amino]benzoate) was only moderately active (MRSA: MBIC 781.25–1562.5 μg/mL, MBEC 1562.5–3125.0 μg/mL; SE: MBIC 781.25–1562.5 μg/mL, MBEC > 1562.5 μg/mL) compared with ciprofloxacin (MRSA: MBIC 0.381 μM, MBEC 48.8 μg/mL, SE: MBIC 0.381–0.7625 μg/mL, MBEC 97.6–195.3 μg/mL). The methyl ester was also the least cytotoxic. Thus, 3,5-dihalogenosalicylic scaffold is essential for antimicrobial activity—iodine atoms preferred (3,5-diiodo, followed by 3-iod-5-chloro- substitution) [93][99]. Simplifying the structure of rafoxanide (a veterinary anthelmintic) by changing the amide group with azomethine and eliminating the phenoxy substituent, an imine analogue, (E)-2-{[(4-chlorobenzyl)imino]methyl}-4,6-diiodophenol (7) was obtained [94][100]. Compound 7 presented selectivity on Gram-positive bacteria, exhibiting antistaphylococcal (MIC 15.625–62.5 μM) and antienterococcal (MIC 62.5–125 μM) activities on reference strains and clinical isolates. The action is bactericidal, and the mechanism indicated inhibition of protein synthesis pathways followed by inhibition of nucleic acid and peptidoglycan production. It exhibits moderate-to-good antibiofilm activity against MRSA and SE (MRSA: MBIC 62.216–124.432 μg/mL, MBEC 124.432–248.863 μg/mL; SE: MBIC 31.108–62.216 μg/mL, MBEC 124.432–248.863 μg/mL) compared with ciprofloxacin (MRSA: MBIC 0.381 μM, MBEC 48.8 μg/mL, SE: MBIC 0.381–0.763 μg/mL, MBEC 97.6–195.3 μg/mL). Due to its bactericidal action, compound 7 seemed to reduce bacterial metabolic activity and inhibit the viability of the released planktonic cells from the biofilm [94][100]. Combining two pharmacophores—salicylaldehyde and sulphonamides—two series of Schiff base analogues of sulfamethoxazole (compounds 8), sulfathiazole (compounds 9) and sulfamethazine (compound 10) were synthesized [95][101]. The influence of the substitution of salicylaldehyde moiety (R2) on antimicrobial activity was investigated. Gram-positive bacteria, especially Staphylococci, were susceptible to the action of the analogues (MIC ≥ 15.62 μM), including clinical isolates (MIC ≥ 3.91 μM) and resistant species (methicillin-resistant S. aureus, MRSA, cotrimoxazole resistant species). Interestingly, the Schiff bases were bactericidal in action compared with sulfonamides and active against cotrimoxazole resistant bacteria, exhibiting no cross-resistance. Eight compounds (8c–d, 9b–d, 10a–c) had MICs (15.62–31.25 μM) comparable to bacitracin (MIC 7.81-15.62 μM) against S. aureus. Once more, the most favourable outcome was achieved with the 3,5-dihalogen substitution on the salicylaldehyde molecule, particularly with the presence of at least one iodine atom, making sulfamethazine derivatives (10) the most potent [95][101]. 4-[(3,5-Dichloro-2-hydroxybenzylidene)amino]-N-(4,6-dimethylpyrimidin-2-yl)benzene-sulfonamide (10a) inhibited MRSA and S. epidermidis biofilm formation (MBIC 390.6–781.25 μM, MBEC > 3462 μM) being inferior to ciprofloxacin (MRSA: MBIC 0.381 μg/mL, MBEC 48.8 μg/mL, SE: MBIC 0.381–0.763 μg/mL, MBEC 97.6–195.3 μg/mL). The compound was not able to disrupt the preformed matrix [95][101]. 5-(4-Methylpiperazin-1-ylsulfonyl)benzylidene)anilines (11a–f) were synthesized and evaluated for antibacterial and anti-Candida actions [96][102]. The antibacterial activity varied among the strains and was influenced by the radical R used. B. subtilis was the most susceptible, followed by P. aeruginosa. Unsubstituted 11a was more potent than the reference (ciprofloxacin—MIC 50 μg/mL) against PA, with electron-donating groups (4-OCH3, 11f) increasing the activity. For S. aureus and E. coli biofilm inhibition, the compounds were inferior to ciprofloxacin. The most favourable substituent was the electron-withdrawing CF3 in ortho or in meta position (11b, 11c). Electron-donating OH seemed essential for antifungal and antibiofilm activity. Derivatives 11d (2-OH), 11c (3-CF3) and 11e (4-OH) surpassed fluconazole (MIC 50.0 μg/mL) in terms of anti-Candida activity. A similar trend was observed for fungal antibiofilm action, with compound 11d (2-OH) being the most active, followed by 11a (H), 11b (3-CF3) and 11e (4-OH) (fluconazole, IC50 40 μM). Compound 11d (2-(2-Ethoxy-5-(4-methylpiperazin-1-ylsulfonyl)benzylideneamino)phenol) inhibited the formation of C. albicans biofilm without affecting planktonic cells, which may indicate a quorum sensing mediated mechanism of action. The docking study against Candida secreted aspartyl protease (SAP5), the enzyme responsible for cell-to-cell adhesion and biofilm formation [97][136], indicated that the 11d is held in place by van der Waals interactions, while 4-methylpiperazine ring form hydrophobic interactions with amino acids at the active site. The azomethine group is also responsible for strong van der Waals hydrophobic and charge bonds interactions with important active site amino acid residues (Ile12, Lys83, Gly85, Asp86, Gly220, Thr221, Thr222, Thr222, Ile223, Tyr225 and Ile305) [96][102]. 4-(o-Methoxyphenyl)-2-aminothiazole was reported to possess antibacterial and antibiofilm potential [98][137]. Its Schiff bases with substituted salicylaldehydes (12a–f) and 2-hydroxy-1-naphtylaldehyde (12g) were synthesized and evaluated for the same effects [99][103]. 4-Bromo-2-(((4-(2-methoxyphenyl)thiazol-2-yl)imino)methyl)phenol (12f) and 2-(((4-(2-methoxyphenyl)thiazol-2-yl)imino)methyl)naphthalen-1-ol (12g) exhibited antibacterial action against B. subtilis (MIC 25 μg/mL). Compound 12g was also active against E. coli (MIC 100 μg/mL) [99][103], with Schiff bases surpassing the parent amine (amine: MIC 250 μg/mL for B. subtilis, 500 μg/mL for E. coli) [98][137]. Regarding antibiofilm potential, compounds 12f and 12g were able to inhibit P. aeruginosa biofilm formation but they do not affect the viability of the cell, suggesting a quorum sensing mechanism of action [99][103]. A series of Schiff bases starting from 2-amino-5-chloro-benzophenone was obtained using microwave irradiation and evaluated for antibiofilm and antibacterial activity [100][104]. Twelve compounds presented MBIC under 100 μg/mL (13a–k). The antibacterial/ antibiofilm activity depended on the type and nature of substituents (R, R1), with electron-donating groups (methoxy, hydroxy) and halogens being favourable. The salicylaldehyde derivative (13d) was only active against S. mutans. The introduction of halogen atoms extended the action to S. aureus (F—13e), K. pneumoniae (Br—13f) and P. mirabilis (Cl, Br—13g). The acridine derivative (13l) and compounds 13a–c inhibited both Gram-positive and Gram-negative bacteria, being inferior to cefixime (MIC 41 μg/mL). S. aureus biofilm was significantly disrupted by compounds 13i, 13k and 13g, while 13i was also active against preformed biofilm of P. mirabilis [100][104]. 4-aminophenazone Schiff bases with different substituted cinnamaldehydes (14a–c) were obtained and tested for antimicrobial and antibiofilm activity. 4-(2-Bromo-3-phenyl-2-propenylideneamino)-1,5-dimethyl-2-phenylpyrazol-3-one (14a) exhibited broad antimicrobial spectrum. It inhibited all fungal strains and all tested bacteria, except P. aeruginosa, exhibiting bactericidal (K. ozaenae, S. enterica) or bacteriostatic effect (E. gergoviae). It reduced up to 90.41% of the biofilm of C. tropicalis and between 75–83% of E. faecalis and S. aureus. Compounds 14b and 14c were also active on biofilm [101][105]. Bacterial fatty acid synthetase may serve as the target for the development of new antibacterial agents. Triclosan and other 2-hydroxydiphenyl ethers demonstrated inhibition against enoyl-acyl carrier protein reductase (FabI), a key enzyme in fatty acid production [102][103][138,139]. Schiff bases and hydrazones have also been reported as inhibitors of staphylococcal β-ketoacyl carrier proteinsynthase III (encoded by FabH gene) [104][105][140,141]. In Gram-negative bacteria, PqsD—an enzyme implicated in Pseudomonas autoinductors synthesis—is structurally related to FabH. Thus, inhibitors of fatty acid synthetases may also act against PqsD [106][142]. Linezolid derived Schiff bases were synthesized starting from 4-(4-amino-2-fluorophenyl)-morpholine in order to obtain PqsD enzyme inhibitors [107][106]. Biofilm inhibition varied according to radical R used. The quinoline derivatives (N-((2-chloroquinolin-3-yl)methylene)-3-fluoro-4-morpholinoaniline—15h, N-((2-chloro-8-methylquinolin-3-yl)methylene)-3-fluoro-4-morpholinoaniline—15i) exhibited the greatest activity against P. aeruginosa biofilm (15h—IC50 12.97 ± 0.33 μM,15i—IC50 15.63 ± 0.20 μM), surpassing linezolid (IC50 15.93 ± 0.18 μM). They also presented good anti-Pseudomonas activity, comparable to linezolid (MIC 2.5 μg/mL) and antimicrobial action against E. coli and B. subtilis (15h, 15i: MIC 5–10 μg/mL, linezolid: MIC 2–3 μg/mL). The docking studies of 15h and 15i against PqsD enzyme revealed van der Waals interactions, hydrophobic bonds (methylene groups of morpholine, methyl group of quinoline) and hydrogen bonds (fluorine atom of linezolid moiety, azomethine group) with amino acid residues of the active site (Figure 4). The phenyl derivatives (15a–e) exhibited only moderate antibiofilm and antimicrobial activities, with electron-donating groups (halogen and methoxy) slightly increasing the effect. Indolyl (15g) and furanyl (15f) derivatives presented no enhancement (Figure 4) [107][106].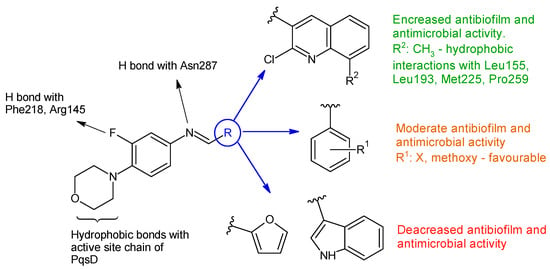
Figure 4.
Structure–activity relationship for compounds
15
, including interactions with PqsD enzyme.
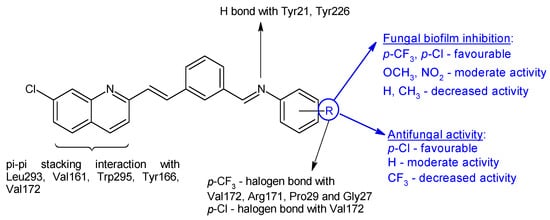
Figure 5.
Structure–activity relationship for compounds
16
, including interactions with Als-3 adhesin of
C. albicans
.
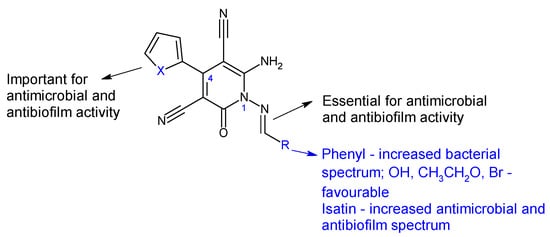
Figure 6.
Structure–activity relationship for compounds
18, 19
.
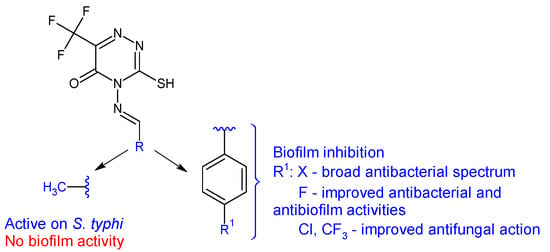
Figure 7.
Structure–activity relationship for compounds
20
.
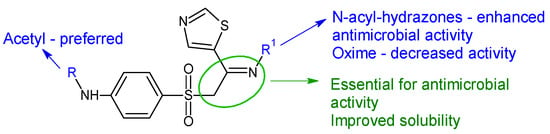
Figure 8.
Structure–activity relationship for compounds
30
,
31
.
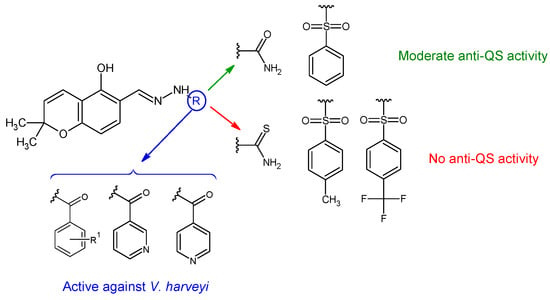
Figure 9.
Structure–activity relationship for compounds
36
.
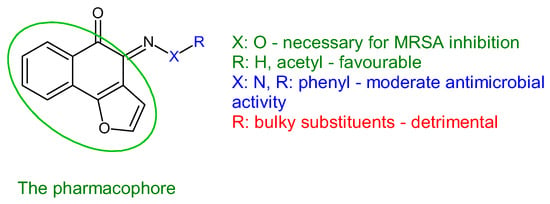
Figure 10.
Structure–activity relationship for compounds
43
.