Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Vivek Kumar Morya and Version 3 by Mona Zou.
Tendinopathy is a debilitating condition marked by degenerative changes in the tendons. Its complex pathophysiology involves intrinsic, extrinsic, and physiological factors. While its intrinsic and extrinsic factors have been extensively studied, the role of physiological factors, such as hypoxia and oxidative stress, remains largely unexplored.
- tendon disorders
- hypoxia
- oxidative stress
- tendinopathy
- degenerative disorders
1. Role of HIF-1 in Tendons
HIF-1 is a crucial regulatory protein that controls oxygen homeostasis and plays a primary role in managing the balance between oxygen supply and demand [1][24]. Under low-oxygen conditions, the hypoxic environment triggers the synthesis of HIF-1, which then regulates angiogenesis, erythropoiesis, and glycolysis to restore oxygen equilibrium [2][25]. To comprehend the oxygen requirements necessary to govern the physical environment of a particular tissue, it is imperative to understand oxygen tension in the tendon [3][4][26,27]. Tendons are highly avascular tissues with oxygen concentrations estimated to be between 1 and 5% [3][5][6][26,28,29]. This limited vasculature can result in hypoxia accompanying tissue damage, making tendinopathy treatment more challenging [7][16]. Research has established that multiple factors contribute to chronic tendon disorders, and among them, HIF-1 may play a role in mediating tendinopathy pathogenesis [7][8][16,30]. Previous studies have shown that HIF-1 is significantly upregulated in tendinopathic tendons, where it regulates the expression of pro-inflammatory cytokines, apoptotic mediators, and angiogenesis, exacerbating tendinopathy [7][8][9][16,30,31].
A well-documented impairment of the HIF-1/vascular endothelial growth factor (VEGF) pathway has been observed in chronic tendon disorders. In tendinopathy, due to elevated hypoxia, HIF-1α expression is upregulated, leading to the promotion of neo-angiogenesis [10][32]. Angiogenesis, mediated by VEGF, plays a crucial role in tendon healing, particularly during acute tendon injuries [8][10][30,32]. However, in chronic tendon injuries, persistently high VEGF expression can result in the formation of hypertrophic scars and keloids, the stimulation of MMPs, and the inhibition of TIMPs [8][10][11][12][30,32,33,34]. Furthermore, hypervascularization may lead to excessive degradation of the ECM, which can be detrimental to tendon health [11][33]. Prolonged VEGF upregulation in a hypoxic environment may lead to excessive scarring, which results in altered ECM homeostasis and excessive ECM degradation, ultimately leading to collagen accumulation [7][11][13][16,33,35]. Based on these factors, thwe researchers sspeculated that hypoxia-induced and VEGF-mediated neo-angiogenesis may play a significant role in tendinopathy pathogenesis [7][8][9][11][13][16,30,31,33,35]. Currently, there is limited research on the role of impaired hypoxia-related genes in chronic tendon disorders. Nonetheless, the available in vitro, in vivo, and clinical studies support the idea that impaired hypoxia persists throughout tendinopathy. Therefore, the regulation of impaired hypoxia may be a valuable strategy for promoting tendon healing.
1.1. HIF-1α Is Degraded under Normoxia
Under normoxic conditions, HIF-1α is constantly synthesized and degraded via ubiquitin-mediated proteolysis [14][36]. However, under hypoxic conditions, HIF-1α rapidly accumulates owing to a decrease in its degradation rate. HIF-1α is hydroxylated by prolyl hydroxylases (PHD1, PHD2, PHD3, and PHD4), which are necessary for its degradation. PHD2 primarily hydroxylates HIF-1α/2α, whereas PHD3 is associated with HIF-2α hydroxylation [14][15][36,37]. Following prolyl hydroxylation, HIF-1α was recognized by the E3 ubiquitin ligase complex formed by the von Hippel–Lindau tumor suppressor protein (pVHL), Elongin B/C, Cullin-2, and RING-box protein 1 (Rbx1). This recognition triggered ubiquitination of HIF-1α by the ligase complex, leading to its proteasomal degradation. (Figure 1) [15][37].
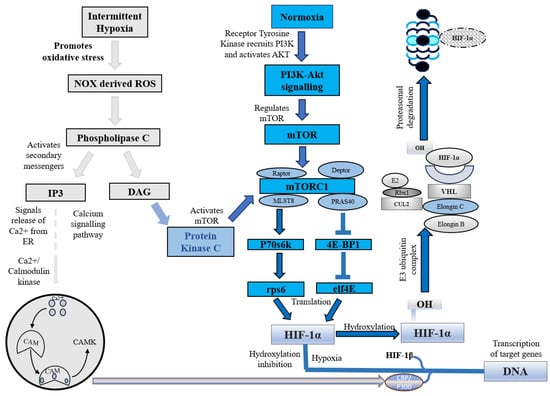
Figure 1. HIF-1α under intermittent hypoxia and normoxia. NOX, NADPH oxidase; ROS, Reactive oxygen species; IP3, Inositol 1,4,5-trisphosphate; DAG, Diacylglycerol; CAM, Calcium/calmodulin; CAMK, Ca2+/calmodulin-dependent protein kinase; mTOR, Mammalian target of rapamycin; PI3K, Phosphoinositide 3 kinase; AKT, Protein kinase B; mTORC1, Mammalian target of rapamycin complex 1; MLST8, Mammalian lethal with SEC13 protein 8; PRAS40, Proline-rich Akt substrate of 40 kDa; P70s6k, Ribosomal protein S6 kinase beta-1; eIF4E, Eukaryotic translation initiation factor 4E; 4E-BP1, Eukaryotic translation initiation factor 4E binding protein 1; rps6, Ribosomal protein S6; HIF-1α, Hypoxia-inducible factor 1-alpha; OH, Hydroxide; CUL2, Cullin-2; Rbx1, Ring-Box 1; E2, Ubiquitin-conjugating enzymes; E3, E3 ubiquitin ligases; VHL, Von Hippel–Lindau.
In addition to this canonical pathway, a non-canonical pathway for HIF-1α inhibition is mediated by a factor that inhibits hypoxia-inducible factor (FIH). Under normoxic conditions, FIH inhibits HIF-1α by hydroxylating an asparagine residue (Asn803 in human HIF-1α and Asn851 in human HIF-2α) within the C-terminal transactivation domains of HIF-1α and 2α. This reduces the activity of the CTAD domain and cooperates with p300/CBP, a histone acetyltransferase [16][38]. However, under hypoxic conditions, the FIH activity is inhibited, allowing HIF-1 to regulate the transactivation of target genes [17][39].
1.2. HIF-1 Expression under Hypoxic Conditions
Hypoxia, a condition characterized by a lack of adequate oxygen supply, is known to play a critical role in regulating both tissue and cell fate. The primary regulator of hypoxia is hypoxia-inducible factor, specifically the constitutively expressed subunit HIF-1β and the oxygen-regulated subunit HIF-1α. In response to hypoxia, HIF-1α is hydroxylated and accumulates in the cytosol before being transported to the nucleus, where it dimerizes with HIF-1β [18][40]. This dimer then binds to hypoxia response elements (HREs) within oxygen-regulated genes [19][41], enabling the activation of genes involved in angiogenesis, erythropoiesis, and the regulation of cell survival, proliferation, and metabolism (Figure 2) [2][19][20][25,41,42]. Therefore, its expression is crucial for the regulation of several key molecular pathways.
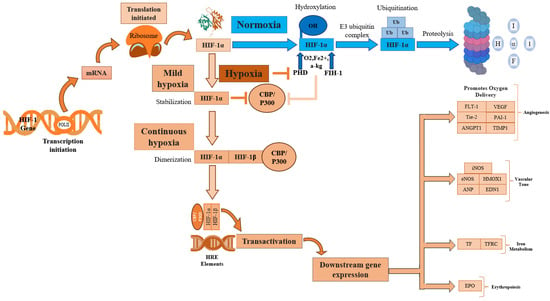
Figure 2. HIF-1α activation regulates several key cellular processes. HIF-1α, Hypoxia-inducible factor 1-alpha; HIF-1β, Hypoxia-inducible factor 1-beta; POLIII, Polymerase 3; CBP/P300, Histone acetyltransferases CBP and p300; p300, E1A binding protein; CBP, CREB-binding protein; mRNA, Messenger RNA; PHD, Prolyl hydroxylase domain; FIH-1, Factor inhibiting hypoxia-1; HRE, Hypoxia response element; Ub, Ubiquitination; OH, Hydroxide; O2, Oxygen; Fe2+, Iron(II) ion; α-KG, α-ketoglutarate; EPO, Erythropoietin; TF, Transferrin; TFRC, Transferrin receptor; iNOS, Inducible nitric oxide synthase; eNOS, Endothelial nitric oxide synthase; HMOX1, Heme Oxygenase; ANP, Atrial natriuretic peptide; END1, Endothelin1; FLT1, Fms related receptor tyrosine kinase 1; VEGF, Vascular endothelial growth factor; ANGPT1, Angiopoietin-1; Tie-2, Angiopoietin-1 Receptor; PAI-1, Plasminogen activator inhibitor 1; TIMP-1, Tissue inhibitor of metalloproteinases.
1.3. Upregulation of HIF-1 in Animal Tendinopathy Models
Animal models of tendinopathy have been proven to be effective in replicating the disease condition. These models show a cellular milieu similar to that observed in tendinopathy. Zhang and colleagues reported that elevated levels of alarmins (produced by injured tissues) were present in a rotator cuff impingement mouse model. Additionally, HIF-1α, an important alarmin, was found to be significantly increased at both the gene and protein levels [21][43]. Sahin et al. developed a rat patellar tendinosis model to investigate the effects of HIF-1α-mediated VEGF angiogenesis on the biomechanical properties of tendons. Researchers have observed that HIF-1α overexpression during tendinopathy promotes VEGF expression. The overexpression of HIF/VEGF may lead to the breakdown of the surrounding tissue through VEGF-regulated MMP release, resulting in a decrease in the tendon’s mechanical strength. Researchers have proposed that regulating neo-angiogenesis may be a potential treatment for tendinopathy [8][30].
One potential strategy to promote tendon repair may be to modulate the levels of HIF-1α following injury (Figure 2) [12][34]. HIF-1α has been found to be upregulated in tendinopathy [7][8][9][16,30,31], and previous studies have suggested that HIF-1α inhibition can alleviate tendinopathy symptoms [12][22][23][34,44,45]. In a recent study conducted by Jiao et al., YC-1, an inhibitor of HIF-1, was evaluated for its potential in promoting tendon repair. These results demonstrate that YC-1 treatment effectively promoted tendon healing by inhibiting HIF-1 and inflammation. Moreover, YC-1 treatment was found to increase the expression of tendon markers Tenascin C (TNC) and Scleraxis (SCX) [22][44]. Similar findings were observed by Wang et al., who found that the HIF-1α levels were elevated in a rat tendinopathy model induced by prostaglandin E2. Their study suggested that the dysregulation of HIF-1/hedgehog signaling may play a role in tendinopathy. Treatment with an HIF inhibitor, Asperosaponin VI (ASA VI), promoted tendon repair via the upregulation of tenogenesis-associated markers such as SCX, TNC, collagen 1, and Mohawk X(MKX), and the downregulation of tendinopathy markers MMP, VEGF-A, and KDR. ASA VI treatment also reduced the expression of Hgg factors, including Shh, Ptch1, Gli1, and Sox 9. Researchers have proposed that in an anaerobic environment, HIF-1α may induce hedgehog signaling, but ASA VI may promote tendon repair by inhibiting HIF-1α and Shh expression in an autocrine manner [23][45]. The positive effects of HIF-1 inhibition in attenuating other musculoskeletal diseases (such as rheumatoid arthritis and osteoarthritis) have been reported [24][25][46,47]. Meng and colleagues evaluated the efficacy of AMSP-30m (a HIF-1α inhibitor) in alleviating arthritic symptoms in an adjuvant-induced arthritis rat model. The results showed that AMSP-30m treatment induced anti-arthritic effects through its anti-angiogenic and anti-inflammatory activity, suggesting that HIF-1α regulation may serve as a treatment for rheumatoid arthritis [24][46]. Additionally, Casticin was found to alleviate inflammation in a knee osteoarthritis rat model. Casticin may improve knee osteoarthritis by inhibiting hypoxia and due to its anti-inflammatory capacity [25][47]. These findings indicate that the proper regulation of HIF-1α may be a viable strategy for the treatment of various musculoskeletal diseases, including tendinopathy.
1.4. Effect of HIF-1 under Differential Hypoxia on Tenocyte/TDSC Proliferation In Vitro
To simulate hypoxic conditions in vitro, researchers have employed various methods, such as the use of chemicals (ascorbic acid or Cobalt Chloride) and hypoxia induction incubators (Anaeropack). These models have the potential to uncover the effects of differential hypoxia on tendon cell populations [3][6][26,29]. Liang et al. evaluated the effects of total hypoxia (0.1%) on human tenocytes. They observed a significant increase in the levels of HIF-1α and hypoxia-induced apoptosis after 48 h. This increase in apoptosis was attributed to the upregulation of the hypoxia-related pro-apoptotic proteins BNIP3 (BCL2/adenovirus E1B 19 kDa protein-interacting protein 3) and NIX (BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like). Researchers have suggested that poor healing of the tendon may be due to increased cellular apoptosis [9][31]. While hypoxia is well documented as inhibiting cell proliferation, it also serves a physiological function for stem cells, including tendon-derived stem cells (TDSCs). TDSCs, the resident stem cell population at the tendon, are considered a viable therapeutic option for the treatment of chronic tendon disorders due to their tenogenic lineage differentiation capacity [26][48].
An in vitro study demonstrated that a low oxygen concentration of 2% promotes the proliferation and lineage differentiation of TDSCs, as opposed to normoxic conditions of 20%. The study also found that TDSCs exhibited higher clonogenicity and proliferative capacity under low oxygen concentrations [27][49]. These findings were supported by another study conducted by Zhang et al., who tested the multi-differentiative potential of TDSCs under different hypoxic conditions. The study found that a low oxygen concentration of 5% was optimal for promoting the self-renewal and differentiation of TDSCs into adipocytes, chondrocytes, and osteocytes. Additionally, researchers observed that hypoxia promotes the expression of stem cell and tenocytic markers [5][6][28,29]. Another study conducted by Yu et al. also reported that a low oxygen concentration of 5% was optimal for promoting the self-renewal capacity of human TDSCs [28][50]. The enhanced proliferative and differentiative capacity under low-oxygen conditions can provide a means to rapidly expand the TDSC population and evaluate the effects of hypoxia in an in vitro model [6][28][29,50].
1.5. HIF-1 Impairment Promotes Tendinopathy
Following tendon injury, there is an increase in the levels of hypoxia-associated genes; therefore, an increase in HIF-1α expression is now considered a biomarker of early tendinopathy [7][16]. HIF-1α overexpression has been shown to induce structural alterations, upregulate apoptotic genes, and promote the expression of inflammatory cytokines, leading to mitochondrial dysfunction, increased calcification, and tissue degeneration [8][9][29][30,31,51].
Józsa and colleagues observed hypoxia-induced structural alterations in the tendinopathic tissues of tendinopathy patients [29][51]. A recent study reported that hypoxia can induce mitochondrial structural alteration, leading to mitochondrial dysfunction [30][52]. Furthermore, hypoxia can promote osteochondrogenic differentiation and tissue calcification, a well-documented feature of tendinopathy [31][53].
As previously mentioned, the overexpression of HIF-1α is well documented in human tendinopathy [7][21][16,43]. Benson and colleagues compared the expression of HIF-1 and BNIP3 in rotator cuff tendinopathy patients against controls and found a marked increase in the levels of HIF-1α and BNIP3 in all patients compared to the controls. This leads to excess cellular apoptosis due to BNIP3 regulation, resulting in poor healing of the tendon [32][17]. In another study, the levels of HIF-1 and VEGF were found to be elevated in samples of rotator cuff tendon disease, and there was also a marked increase in the levels of MMPs, which promotes matrix degradation [33][54]. Mosca et al. compared the levels of HIF-1α pre- and post-tendinopathy treatment and found that the levels of HIF-1α were increased in diseased tendons when compared to post-treatment and healthy tendons [34][55]. Millar and colleagues also reported an increase in HIF-1α levels in tendinopathy samples, which may promote the expression of pro-inflammatory cytokines and various MMPs, disturbing ECM homeostasis and leading to degeneration [7][16]. These studies suggest that an impaired HIF-1-mediated pathway may contribute to tendon degeneration, and targeting the components of this pathway may be a viable treatment for tendon-related disorders.
1.6. Regulating the Hypoxic Environment May Promote Tendon Repair
Because a hypoxic environment is a physiological condition for various stem cell niches, including embryonic and adult somatic stem cells, researchers have utilized hypoxic preconditioning to promote the proliferative and differentiative capacity of various stem cells, including mesenchymal stem cells (MSCs), adipose-derived mesenchymal stem cells (AD-MSCs), bone-marrow-derived stem cells (BMSCs), and TDSCs [6][27][28][35][29,49,50,56]. These studies have documented that hypoxic preconditioning also promotes the expression of tenogenic markers in these stem cells [36][37][38][57,58,59]. The transplantation of these stem cells, particularly those pre-conditioned under hypoxic conditions, has been reported to be effective in treating chronic tendon disorders. For example, a study found that hypoxia-preconditioned BMSCs showed improved histological scores and biomechanical properties in a rabbit tendon injury model compared with normoxic BMSCs [36][57]. Another study expanded upon the previous findings to determine how transplanted cells migrated and functioned following engrafting. Hypoxic MSCs labeled with super magnetic iron oxide were found to migrate into the punch area of the supraspinatus tendon and engraft into the injured tendon. The labeled cells were detec1even four weeks post-transplantation and significantly improved biomechanical strength and enhanced recovery in a rat rotator cuff tear model [39][60]. Thus, hypoxic pre-conditioning plays an important role in mediating stem cell differentiation, and the transplantation of hypoxic stem cells significantly improves the histological score and biomechanical properties [27][37][38][39][49,58,59,60]. However, the exact mechanism by which hypoxia mediates differentiation potential remains unknown [36][57], and further research is needed to determine whether it functions as an inducer of other growth factors.
Unfortunately, tendinopathy is a complex, multifactorial disorder with an uncertain etiology [40][61]. However, the increased expression of the hypoxia marker HIF-1 following tendon injury may play a critical role in its initiation [7][16]. ThWe researchers sspeculate that regulating the expression of HIF-1 and its target genes, as shown in Figure 3, may help to attenuate tendinopathy severity. One possible mechanism by which HIF-1 may contribute to tendinopathy is the regulation of MMP3 by its activator TIMP-1 [41][62]. Exposure to hypoxia increases HIF-1α levels, which in turn activates several key pathways, including MMPs and their inhibitors, TIMPs. TIMP-1, in particular, plays an important role in regulating MMP levels, as a balance of MMP/TIMP is necessary for maintaining tendon homeostasis. An imbalanced MMP/TIMP ratio caused by dysregulated TIMP-mediated MMP regulation may lead to impaired remodeling during tendon repair [42][43][63,64]. TIMP-1 is known to play a role in various biological processes, including tissue remodeling, promoting growth factor activity, and wound healing [43][64]. As an MMP inhibitor, TIMP-1 primarily targets and inhibits MMP-9. However, it also has a strong affinity for MMP-3. The MMP3–TIMP1 complex is formed by interactions between the catalytic domains of human stromelysin-1 (MMP-3) and human TIMP-1 [44][45][65,66]. Further research is necessary to fully understand the role of HIF-1 and TIMP-1 in tendinopathy and to develop effective treatments for this debilitating condition [42][45][63,66].
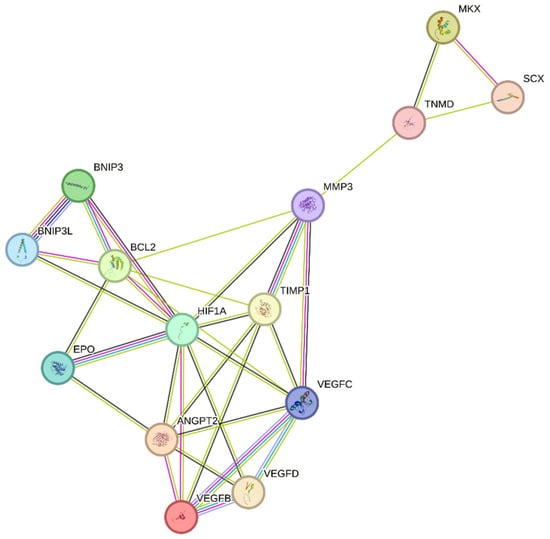
Figure 3. Regulation of the hypoxia factor HIF-1α may promote tendon repair in chronic tendon disorders. HIF-1α, Hypoxia-inducible factor 1-alpha; BCL2, B-cell lymphoma/leukemia-2; SCX, Scleraxis; MMP-3, Matrix Metalloproteinase 3; MKX, Mohawk X; EPO, Erythropoietin; ANGPT2, Angiopoietin 2; VEGFD, Vascular endothelial growth factor D; VEGFC, Vascular endothelial growth factor C; VEGFB, Vascular endothelial growth factor B; BNIP3, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3; BNIP3L, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like; TIMP1, Tissue inhibitor of metalloproteinases 1; TNMD, Tenomodulin.
Previous research has indicated that TIMP-1 hinders excessive matrix degradation by impeding MMP activity, especially that of MMP-9, as MMP-2 and MMP-9 are upregulated in tendinopathies [42][63]. Consequently, the inhibition of MMP-2 and MMP-9 by their respective TIMPs can promote tendon repair. In addition to MMP-2 and MMP-9, MMP-3 has been found to be dysregulated during tendinopathies, which is inconsistent with its expected role in matrix remodeling. A study also suggested that the expression of MMP-3 is suppressed during tendinopathy, and MMP-3 polymorphism may contribute to chronic tendon disorders [46][67]. As MMP-3 also plays a crucial role in matrix remodeling, the downregulation or complete inhibition of MMP-3 by TIMP-1 would imply the failure of proper matrix remodeling [47][68]. ThWe researchers sspeculate that following tendon rupture, elevated HIF-1α increases TIMP-1 expression, which in turn inhibits MMP-3. This inhibition of MMP-3 may lead to the altered expression of tenocytic markers, such as SCX, MKX, and TNMD. This suggests that MMP-3 may be involved in the maintenance of tenocytic homeostasis. Therefore, a possible treatment strategy may be to balance the regulation of the TIMP-1/MMP-3 complex. Further studies focusing on the regulation of this complex following tendon injury may be a viable treatment strategy to promote tendon repair and may lead to increased expression of tenocytic proliferative markers (SCX, MKX, and TNMD) (Figure 3).
Recently, the hypoxia-associated apoptotic gene BNIP3 has been linked to tendinopathy [9][32][17,31]. A study of patients with subacromial impingement and rotator cuff tears revealed a significant increase in HIF-1α levels. This overexpression leads to the buildup of fragmented DNA in tissue samples, suggesting that HIF-1α promotes apoptosis in hypoxic environments. Researchers have proposed that the upregulation of the apoptotic gene BNIP3 leads to apoptosis of fibroblasts and fibroblast-like cells, resulting in reduced collagen synthesis and a hypoxic environment [32][17]. Therefore, maintaining a dynamic balance between pro-apoptotic and anti-apoptotic factors in a hypoxic environment is crucial for effective tissue healing [32][48][17,69]. This is further supported by another study, which found that cells lacking HIF-1 showed a reduced expression of BNIP3 and underwent lower apoptosis, indicating that BNIP3 is a direct target of HIF-1α [49][70]. The apoptosis associated with tendinopathy results in reduced cell proliferation and increased chondrogenic metaplasia, a hallmark of tendinopathy [9][32][17,31]. The manipulation of BNIP3 expression following HIF-1α upregulation may be a potential treatment for tendinopathy (Figure 3).
In recent years, VEGF has garnered considerable attention as a potential therapeutic target for promoting tendon repair. Following tendon injury, hypoxia induces VEGF expression, thereby promoting increased vascularization [8][10][30,32]. However, in tendinopathy, VEGF expression remains elevated during the remodeling phase, impeding the tendon repair process. This prolonged VEGF-induced angiogenesis leads to the upregulation of matrix metalloproteinases (MMPs) and the suppression of tissue inhibitors of metalloproteinases (TIMPs), resulting in extracellular matrix (ECM) degradation and collagen 1 downregulation, which negatively impact tendon repair [8][50][30,71]. Moreover, in a fully healed tendon, the blood vessels usually regress; however, in tendinopathy, excessive blood vessels resulting from prolonged VEGF overexpression impair the tendon’s biomechanical properties, promote scar tissue formation and inflammation, and delay the healing response [8][51][52][30,72,73]. In the context of tendon repair, the overexpression of VEGF can result in poor and abnormal vasculature, commonly referred to as “hyperpermeable”. These vessels are leaky and incapable of providing adequate oxygen and nutrients, thereby hindering proper tissue regeneration [53][74]. The prolonged presence of blood vessels and high levels of VEGF can also lead to the upregulation of MMPs and scar formation, further impairing tendon repair [11][33].
To facilitate tendon repair, it is crucial to regulate angiogenesis and VEGF at an early stage [8][10][30,32]. The presence of blood vessels and high levels of VEGF for extended periods can have detrimental effects [8][51][30,72]. Therefore, an alternative strategy is to stabilize the neovessels, which can provide adequate oxygen and nutrients to support tendon regeneration [53][74].
Tendon injury leads to the production of HIF-1α, resulting in the overexpression of VEGF [50][52][71,73]. While this process can aid in tendon repair, in the case of chronic tendon disorders, prolonged angiogenesis can weaken the mechanical strength of the tendon by breaking down the surrounding tissue through VEGF-triggered MMPs and upregulation of the hypoxia-associated apoptotic gene BNIP3. This leads to poor matrix remodeling and cell death [8][9][32][17,30,31]. Studies have demonstrated that the regulation of hypoxia can promote tendon repair. Tendon repair can be enhanced by regulating the HIF-1α/VEGF/BNIP3-mediated pathway (Figure 3).
2. Hypoxia and Oxidative Stress Play an Important Role in the Pathogenesis of Tendinopathy
2.1. Hypoxia Regulates Oxidative Stress in Chronic Tendon Disorders
In conclusion, hypoxia and oxidative stress play a crucial role in the development and progression of tendinopathy [7][54][16,23]. The cumulative effects of these factors on tendon degeneration remain unclear. In hypoxic conditions, HIF-1 may modify the cytochrome chain, which is responsible for mitochondrial oxidative phosphorylation, resulting in reduced ATP synthesis and excess ROS formation [7][55][16,138]. This is supported by the fact that hypoxia leads to ROS production, which inhibits PHD activity, resulting in the stabilization of HIF-1α levels [56][139]. The accumulation of HIF-1α under hypoxia may be the result of the inhibition of antioxidants, which would lead to a loss of antioxidant capacity to scavenge excess ROS, thereby inducing systemic and local activation of HIF-1α. HIF-1α, in turn, activates target genes, such as erythropoietin and VEGF, which promote the production of ROS. Chronic hypoxia, via the accumulation of HIF-1α, inadvertently promotes NADPH oxidase activation, which intensifies ROS export and, in turn, promotes oxidative stress [57][58][140,141]. Following tendon injury, the upregulation of HIF-1α leads to excess ROS production, which results in incomplete tendon repair. Although the precise pathogenesis is not yet fully understood, it is speculated that the HIF-1α/ROS cascade may promote the progression of chronic tendon disorders by regulating several pathways [7][8][59][16,30,142]. ThWe researchers propose that HIF-1α regulates oxidative stress via the dysregulation of ROS and, in turn, the promotion of VEGF and other aforementioned factors. This cascade of mechanisms negatively affects tendon health and delays healing (Figure 4).
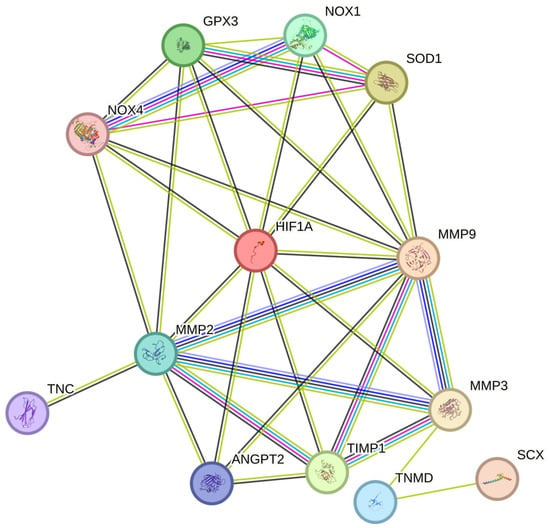
Figure 4. HIF-1α regulates ROS and worsens tendon health during TP. HIF-1α, Hypoxia-inducible factor 1-alpha; MMP-3, Matrix Metalloproteinase 3; SCX, Scleraxis; TNMD, Tenomodulin; TIMP1, Tissue inhibitor of metalloproteinases 1; ANGPT2, Angiopoietin 2; MMP-9, Matrix Metalloproteinase 9; MMP-2, Matrix Metalloproteinase 2; TNC, Tenascin C; NOX1, NADPH Oxidase 1; NOX4, NADPH Oxidase 4; SOD1, Superoxide dismutase 1; GPx3, Glutathione Peroxidase 3.
2.2. Chronic Hypoxia-Induced Oxidative Stress Delays Healing by Prolonging the Inflammatory Response
Chronic hypoxia-induced oxidative stress may impede tendon healing by disrupting the regulation of the NF-κB pathway [60][61][132,143]. NF-κB plays a crucial role in controlling cellular processes, including inflammation, cell survival, and stress response. Previous studies have indicated that NF-κB signaling is elevated in patients with rotator cuff tendinopathy. This increased NF-κB level plays a vital role in degenerative changes in the tendon [60][132]. Hypoxia-associated reactive oxygen species (ROS) cause an increase in NF-κB-associated inflammatory markers, such as TNFa, IL-1B, and PGE2. This inflammatory response is crucial for the removal of necrotic material and the production of collagen to promote tendon healing. However, a prolonged inflammatory response leads to tendon adhesion, which impairs the tendon repair process and negatively affects the tissue-resident cells [61][143]. Although the exact mechanism is still not fully understood, it is believed that the oxidative stress resulting from hypoxia activates NF-κB [62][144]. Under hypoxia, the rate of IKK degradation by IκB increases, leading to the activation of NF-κB and its translocation to the nucleus at a higher rate, where it upregulates inflammatory gene expression. Activated NF-κB further promotes HIF-1 levels, thereby enhancing HIF-1-mediated activity [63][145]. HIF-1, in turn, promotes the expression of NF-κB target genes, such as COX-2 [64][146] and IL-6 [65][147], resulting in a prolonged state of inflammation and delayed healing [66][105].
2.3. Hypoxia Promotes Oxidative Stress by Upregulating NOX and Downregulating Antioxidants
Following tendon injury, the expression of HIF-1 is promoted by hypoxia, which governs several metabolic processes, including oxygen homeostasis, energy metabolism, growth, and differentiation [67][148]. HIF-1 has been documented to promote the expression of the NOX family, particularly NOX1 and NOX4, which are highly elevated in oxidative-stress-induced tendinopathy animal models [68][69][70][92,96,149]. This suggests that a hypoxic environment promotes the expression of NOX members during tendinopathy.
Under typical physiological conditions, the reactive oxygen species (ROS) generated by NOX1 and other NOX isoforms regulate cell growth, differentiation, survival, apoptosis, metabolism, and migration by targeting the redox-sensitive cysteine residues in cellular molecules. Protein tyrosine phosphorylation (PTP) is a common molecule targeted by ROS within cells that controls the phosphorylation of numerous proteins involved in cellular signal transduction, potentially disrupting normal cellular homeostasis. Although the precise mechanism remains unclear, the mitogen-activated protein kinase system and phosphoinositide 3-kinase, which are regulated by the NOX family, including NOX1, have been shown to be activated by NOX4 in multiple studies [70][71][149,150].
The production of NOX4 has been documented to increase under hypoxia because of the autocrine activation of its mediator, transforming growth factor beta-1 [72][151]. Hypoxia promotes oxidative stress by stimulating NOX4 and ROS generation. NOX4 has also been shown to play a role in the chondro-osteogenic phenotype observed in tendon cells during tendinopathy. NOX4 activation under hypoxia promotes ROS expression, which then activates the ERK/JNK pathway, resulting in increased expression of SOX9 (chondrogenic marker) and RUNX2 (osteogenic marker). This indicates that NOX4-derived ROS under hypoxia play a role in the pathogenesis of tendinopathy [73][152] (Figure 4).
NOX1 expression has also been reported to be elevated during hypoxia. The upregulation of NOX1 is associated with an increase in HIF-1α and the stimulation of HIF-dependent target gene transcription. Consequently, an accumulation of H2O2 occurs in cells stably transfected by NOX1, which is believed to be a result of NADPH-dependent superoxide formation and HIF-1α activation [74][153].
Glutathione peroxidase 3 (GPx3) is a potent antioxidant that scavenges ROS and is a major contributor to the reduction of H2O2, hydrogen peroxides, and other oxidants. Elevated GPx3 expression has been documented to alleviate oxidative stress in tendinopathy, suggesting its potential as a therapeutic approach to treating this disorder [75][154]. Furthermore, GPx3 has a binding site for HIF-1, and its expression is regulated by HIF-1 following hypoxic exposure. However, owing to the accumulation of ROS resulting from NOX isoforms, GPx3 levels decrease over time, leading to a deficiency in antioxidants [76][155].
Superoxide dismutase 1 (SOD1) plays a crucial role in protecting cells from the detrimental effects of ROS, particularly superoxide anions. SOD1 specifically targets and neutralizes superoxide radicals by catalyzing the dismutation of two superoxide molecules into molecular oxygen (O2 and H2O2) [77][156]. The enzyme activity relies on a specific catalytic metal ion, which can be manganese (MnSOD), iron (FeSOD), nickel (NiSOD), or copper (Cu/ZnSOD). In eukaryotes, MnSOD (SOD2) and Cu/ZnSOD (SOD1) are documented isoforms [78][157]. Moderate overexpression of SOD reduces local hypoxia and prevents the accumulation of HIF-1. However, decreasing the levels of SOD via siRNA transfection resulted in an increase in the levels of superoxide ions and the accumulation of HIF-1α, which was reversed by using superoxide scavengers such as SOD [79][158]. In the case of chronic hypoxia, the ROS released by NOX may result in excess HIF-1α and ROS accumulation [80][81][159,160], resulting in the suppressed activity of SOD isoforms and the overall induction of a state of oxidative stress mediated by intermittent hypoxia.
Therefore, chronic hypoxia may promote oxidative stress (OS) and lead to poor tendon healing (Figure 4) [55][82][128,138]. Although the exact pathway is unknown, thwe researchers sspeculate that the NOX family [68][69][82][92,96,128], which has been previously reported to regulate the levels of various MMPs [83][161], may delay tendon healing by increasing the levels of tendinopathy-associated MMPs such as MMP9 and MMP2 [83][84][85][161,162,163]. During tendinopathy, hypoxia promotes the expression of HIF-1α, NOX1, and NOX4. The overexpression of NOX isoforms results in the reduced expression of antioxidants such as SOD1 and GPx3, which inadvertently leads to excess ROS accumulation by the NOX family [73][74][75][86][87][152,153,154,164,165]. This, in turn, promotes the expression of various MMPs such as MMP2 and MMP9, and the downregulation of the MMP inhibitor TIMP1, resulting in excess ECM degradation [8][57][58][59][30,140,141,142]. Overall, the tendon repair process was impaired. (Figure 4)
Therefore, correction or partial modulation of the hypoxic response may be a feasible treatment approach to chronic tendon disorders. The regulation of HIF-1 would aid in maintaining the oxidant–antioxidant balance and promoting tendon repair by downregulating MMPs and ROS and ensuring appropriate ECM remodeling (Figure 4).