Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Lindsay Dong and Version 2 by Lindsay Dong.
Despite its involvement in several human pathophysiological processes, the cellular prion protein (PrPC) remains enigmatic. During the last ten years, PrPC has also been reported to be implicated in several human cancers, the molecular mechanisms of which are under investigation. In some tumors, elevated expression of PrPC protein is associated with poor patient prognosis. At the cellular level, high PrPC expression in tumor cells is associated with the acquisition of stemness1-like characteristics, metastatic and invasive process, and resistance to chemotherapy.
- prion protein
- cancer
- drug resistance
- therapeutic target
1. Introduction
1.1. Background
Cellular prion protein (PrPC) is a glycosylphosphatidylinositol (GPI)-anchored glycoprotein expressed on the cell surface in various organs and tissues [1]. The PrPC protein is encoded by the PRNP gene that is localized on Chromosome 20 and 2 in humans and in mice, respectively [1]. PrPC is first synthesized as a pre-pro-protein with a leader peptide at the N-terminal tail, and a GPI anchor signaling peptide (GPI-PSS) at the C-terminal tail. The leader peptide guides the pre-pro-PrPC into the endoplasmic reticulum (ER) where it is cleaved to generate the pro-PrPC (Figure 1). Like other GPI-anchored proteins, pro-PrPC is then translocated from the ER to the Golgi with the help of post-GPI attachment proteins 1 and 5 (PGAP1 and PGAP5) [2][3]. In this compartment, PrPC becomes a mature protein by undergoing further processes such as N-linked glycosylation, GPI-PSS removal, and addition of the pre-assembled GPI anchor [4][5] (Figure 1). PrPC is translocated from the Golgi to the outer leaflet of the plasma membrane where it is inserted via its GPI domain [4][5] (Figure 1).
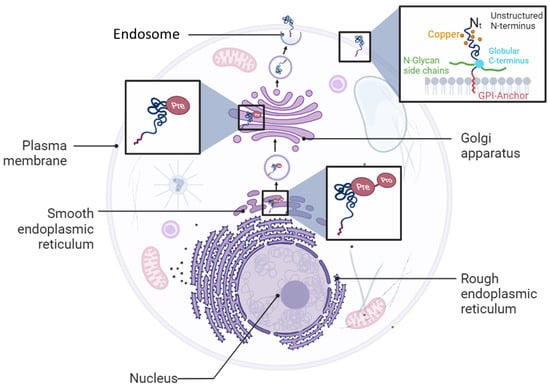
Figure 1. Cellular biosynthetic pathway of PrPC protein. PrPC is synthetized as a pro-pre-protein in the endoplasmic reticulum compartment before trafficking to the Golgi apparatus and plasma membrane where it is anchored as a glycosylphosphatidylinositol (GPI) protein.
PrPC contains a flexible N-terminal domain (Nt) located between residues 23 and 124. It comprises five repetitive motifs of eight amino acids (PHGGGWGQ) that exhibit a high affinity for copper ions (Cu2+). This binding takes place within the HGGGW residues that showed, in vitro, more affinity for Cu2+ than for Cu+ or to any other metal ion [6] (Figure 1 and Figure 2). PrPC also exhibits a globular C-terminal domain (Ct), anchored to the plasma membrane, of about 100 amino acids, from residues 125 to 228. This domain is composed of three α-helices, corresponding to amino acids 144–154, 173–194, and 200–228, interspersed by two antiparallel β sheets of residues 128–131 and 161–164 (Figure 2).
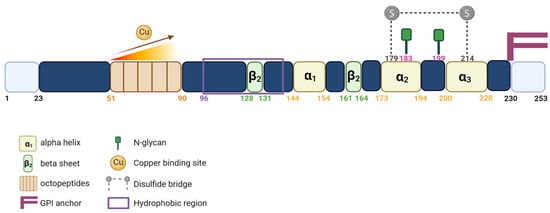
Figure 2. Schematic representation of the PrPC protein. Linear representation of the primary sequence of human PrPC showing important protein domains.
The third α helix and the second β sheet are connected by a flexible loop. There are two N-glycosylation sites (residues 183 and 199), which might not be partially or fully glycosylated, resulting in three distinct forms of the PrPC: the non-glycosylated (~25 kDa), the mono-glycosylated (~25 to 30 kDa) and the bi-glycosylated forms (~35 kDa), respectively. PrPC is also characterized by the presence of a single disulfide bridge between the two cysteine residues 179 and 214, which allows the link between helix 2 and 3 and serves to stabilize the tertiary structure of the PrPC (Figure 2).
1.2. PrPC Expression and Functions
The expression of PrPC begins at embryogenesis [1]. The highest level of PrPC expression was found in the central and peripheral nervous systems [1]. In adults, strong expressions were detected in the brain, spinal cord, neurons, and glial cells [7][8]. PrPC expression was also ubiquitously detected in various cells of the peripheral tissues [9][10][11][12]. The interest in the study of PrPC was mainly related to its incrimination in the pathogenesis of the neurodegenerative disorders known as spongiform encephalopathies (SE) or prion diseases [13][14]. The SE was mainly associated with bovines (BES), and commonly referred to as mad cow disease, which refers to an untreatable and inevitably fatal neurodegenerative illness that affects cattle [15]. BES is characterized by the aggregation of an abnormal beta-sheet rich isoform of the PrPC protein called scrapie (PrPSc) [13][14]. In humans, the corresponding form of BES is Creutzfeldt-Jakob’s Disease (CJD), which is also characterized as a brain degenerative disorder [16]. Fatal familial insomnia (FFI) stands as an exceptionally rare prion disease that induces neurodegeneration and primarily manifests through insomnia, making it incredibly challenging to sleep. The predominant instances of this condition are hereditary in nature, resulting from a mutation in the PRNP gene, while sporadic cases make up the remaining occurrences [17]. Scrapie gives rise to a lethal degenerative ailment that targets the nervous systems of goats and sheep. Classified as one of several transmissible spongiform encephalopathies (TSEs), it is believed to stem from prions [18]. In the last decade, PrPC has also been shown to play a significant role in cancer biology. PrPC has been found to be upregulated or ectopically expressed in different types of cancer tissues, such as hepatocellular carcinoma, gastric cancer, melanoma, breast cancer, colorectal cancer, pancreatic ductal adenocarcinoma, prostate cancer, osteosarcoma, and glioblastoma [19][20][21][22][23][24][25][26][27][28]. The increased expression of PrPC appears to play a crucial role in cancer growth, development, differentiation, invasion, migration, metastasis, chemotherapy resistance, and resistance to apoptosis [19][20][21][22][23][24][25][26]. The growing body of evidence linking PrPC to cancer has opened up new avenues for cancer research [29][30]. The interaction of PrPC with various proteins and receptors leads to the activation of intracellular signaling pathways that promote tumorigenesis [31][32]. The differential expression of PrPC in various types of cancer, its involvement in protein–protein interactions and its activation of downstream pathways confers to this protein a likely role in cancer.32. PrPC in Human Cancers
32.1. PrPC and Gastric Cancer
The expression of PrPC has been reported to be highly elevated in gastric cancer tissue, indicating its potential involvement in the pathogenesis of this disease [20]. Moreover, PrPC has been shown to promote multidrug resistance in gastric cancer cells by inhibiting apoptosis [20]. Due to its ability to bind to certain extracellular matrix and adhesive proteins, PrPC exhibits an adhesive feature, indicating its involvement in cell adhesion [22][26][33]. A comparison of the PrPC expression in primary and metastatic sites was conducted in patients with metastatic and non-metastatic gastric cancer [23]. Although no significant difference in the PrPC expression was observed between the primary and metastatic sites, a higher staining score for PrPC was observed in the metastatic compared to the non-metastatic cancers, indicating a potential correlation between the PrPC expression and gastric cancer aggressiveness.32.2. PrPC and Melanoma
Previous studies have shown that PrPC interacts with Filamin A (FLNA) to promote cancer progression [34]. PrPC-silenced FLNA-deficient M2 melanoma cells exhibited decreased M2 cell migration in wound healing assays [35]. This was further reversed by reintroducing PrPC in PRNP-null M2 cells [36]. Despite the fact that PrPC enhances cell migration and alters the cell cytoskeleton organization through FLNA disruption, M2 cells do not express FLNA. Indeed, the effect of PRNP deletion on cell migration was shown to be associated with F-actin protein. The latter, in wild-type M2 cells that are characterized with a higher mobility, shows an expression level which varies according to that of PrPC [36]. These findings demonstrate that PrPC negatively regulates F-actin without binding to FLNA. To determine the pathway through which PrPC affects F-actin, Hsp27 was assessed based on its importance for cell motility and its ability to reduce actin aggregation [37][38]. The levels and phosphorylation of Hsp27 were evaluated in the presence or absence of PrPC. There was a significant decrease in phosphorylated Hsp27 at Ser82 when PRNP was deleted, and P-Hsp27 levels were rescued when PRNP was re-expressed in PRNP-null M2 cells [36]. To identify the kinase responsible for this observation, the inhibition of P38MAPK, Akt, PKD, PKA, and PKC was assessed, as these kinases have been reported to act on Hsp27 [39][40][41][42][43]. Only Akt inhibition decreased the P-Hsp27 levels that were also decreased when PRNP was silenced. The Akt expression was rescued when PrPC was re-expressed. The binding between Akt and Hsp27 was confirmed by co-immunoprecipitation and co-purification in the presence of PrPC and was higher in comparison to their binding in PrPC-null M2 cells [36]. These findings support the correlation between PrPC and Akt levels, which will disturb the downstream Akt/Hsp27 interaction, inducing the regulation of actin polymerization and cell migration. PrPC interaction with FLNA also promotes FLNA interaction with β1 integrin, contributing to melanomagenesis [44][45]. A7 cells, which express FLNA, exhibited higher spreading and migration ability compared to M2 cells that do not express FLNA [46]. PrPC exists as Pro- PrPC in both A7 and M2 cells, retaining its glycosylphosphatidylinositol anchor peptide signal sequence (GPI-PSS) with an FLNA binding motif.32.3. PrPC and Breast Cancer
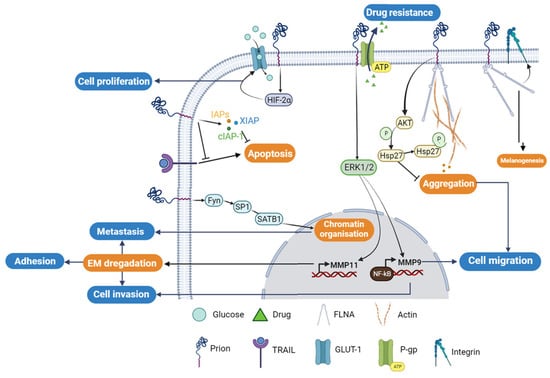
The resistance of cancer cells to apoptosis or drug treatment is one of the main features of tumorigenesis. Epigenetic modifications [47], ectopic gene expression [48][49][50], and oncogene overexpression can lead to aberrant expression of anti- or pro-apoptotic proteins. In breast cancer, PrPC has been reported to contribute to cancer resistance to apoptosis and drug treatment. Chemotherapy of TNF-resistant breast carcinoma cells was effective in patients who were PrPC-negative. However, PrPC overexpression in estrogen receptor (ER)-negative breast cancer patients was linked to decreased sensitivity to chemotherapy, indicating that PrPC could potentially be used as a predictor of adjuvant chemotherapy benefit in ER-negative patients [48]. Overexpression of PrPC has also been shown to cause resistance to TRAIL (Tumor necrosis factor-Related Apoptosis Inducing Ligand)-induced apoptosis in Adriamycin (MCF7/ADR) [51][52][53][54][55][56]. The elevated expression of PrPC in MCF7/ADR and 2101 cell lines compared to MCF7 cells correlates with the breast carcinoma cells’ resistance to Adriamycin and TRAIL-induced cell death [57]. Nevertheless, the knockdown of PrPC using the siRNA-PrPC strategy in resistant cell lines only restores sensitivity to TRAIL-mediated apoptosis by up to 25% in MCF7/ADR and 60% in 2101 cells. This is achieved through the enhancement of Bid cleavage and caspase-3 processing, concomitantly with Mcl-1 downregulation and activation of pro-apoptotic Bax through the downregulation of Bcl-2 [57]. In addition to its role in acquiring resistance, PrPC has been shown to be a crucial factor for invasion and migration of MCF7 breast cancer cells. PrPC overexpression increases matrix metalloprotease-9 (MMP-9) expression by enhancing the association of NF-κB with the promoter of the MMP-9 gene and ERK signaling, similar to that observed in gastric cancer [58] (Figure 3).Figure 3. Cellular regulatory pathways involving PrPC protein in cancer. The PrPC protein is involved in different cellular tumorigenesis process, where its activities are regulated directly through protein–protein interactions or/and indirectly using different cellular regulatory pathways. P-gp; P-glycoprotein, GLUT-1; Glucose transporter-1; FLNA; Filamin A.
32.4. PrPC and Colorectal Cancer
Colorectal adenocarcinoma (CRC) cells exhibit high levels of expression of PrPC compared to normal colorectal cells. PrPC plays a crucial role in tumor growth and survival by promoting the Warburg effect, which involves increased reliance on glucose metabolism, in the presence of oxygen. This process ensures rapid proliferation and survival of cancer cells [59][60]. Through the Fyn-HIF-2α pathway, PrPC increases the expression of GLUT-1, the main glucose transporter, thereby enhancing the dependency of CRC cells on the glycolytic pathway for tumor growth (Figure 3). In contrast, the depletion of PrPC suppresses glucose utilization by suppressing GLUT-1 expression, leading to the inhibition of tumor growth both in vitro and in vivo [61]. Cell surface proteomics studies have identified the differential expression of GLUT-1 and PrPC as potential biomarkers of colorectal adenoma to carcinoma progression. Hence, these proteins can serve as potential targets for the emerging molecular imaging modalities [62]. Functional assays have revealed a molecular mechanism that links the levels of PrPC expression to the regulation of CRC metastasis. Ectopic PrPC expression was found to promote the in vitro metastatic potential of CRC cells, while inhibition of PrPC significantly reduced cancer cell motility [63]. The pathway involving PrPC-mediated upregulation of SATB1 is a matrix attachment region-binding protein that regulates higher-order chromatin organization and tissue-specific gene expression.
In colorectal and pancreatic ductal adenocarcinoma (PDAC), the overexpression of PrPC has been shown to confer resistance to anti-cancer drugs, including doxorubicin, etoposide, and vincristine sulfate [64][65][66][67]. In LS-174T cells overexpressing PrPC, a higher cell viability and less apoptosis were observed compared to non-transfected cells. The PrPC anti-apoptotic effect is thought to be mediated through the upregulation of the three proteins that are involved in the inhibition of apoptotic pathway. These include the inhibitor of apoptosis proteins (IAPs)-survivin, the X-linked inhibitor of apoptosis protein (XIAP), and the cellular inhibitor of apoptosis protein-1 (cIAP-1) [68] (Figure 3).
At the molecular level, it was proposed that the PrPC involvement in PDAC is mediated upon its interaction with filamin A (FLNA). This interaction affects the cytoskeleton organization and the expression of different signaling proteins, triggering the cellular proliferation and invasiveness, leading to overall tumor growth [64][66] (Figure 3).
43. The Potential Diagnostic and Therapeutic Value of PrPC in Different Types of Cancer
PrPC expression has been investigated in various types of cancer, including bladder and prostate cancer, osteosarcoma, and glioblastoma [30][69][70][71][72]. In prostate spheroids, PrPC expression was inversely correlated with the spheroid diameter and related to the intracellular redox state, potentially by contributing to anti-oxidative defense. Moreover, PrPC was found to be overexpressed in 90% of prostate cancer biopsies, although its diagnostic or prognostic value remains unknown [69]. In osteosarcoma, the most common bone malignancy, PrPC was differentially overexpressed and appeared to be associated with tumor development and aggressiveness, as well as a negative regulator of apoptosis [70]. PrPC expression was directly correlated with the proliferation of glioma stem cells (GSC), and its downregulation reduced GSC stemness, cell growth, clonogenicity, and spherogenicity, as well as the ability to develop tumors in animal models. The results imply that PrPC plays a crucial role in preserving GSC stemness [73][74]. Hence, blocking its activity could enhance the sensitivity of cancer cells to chemotherapy [71][75]. Interestingly, PrPC expression was found to increase the sensitivity to doxorubicin in MDA-MB-435 breast cancer cells, unlike colorectal cancer, suggesting a tumor type-specific mechanism [76]. Recent studies have also shown that PrPC is expressed in human lung epithelial cells and is involved in anti-oxidative defense and the maintenance of tight junctions in the epithelial barrier [77].54. Targeting PrPC Interactions in Cancer: New Insights and Potential Strategies (Figure 4)
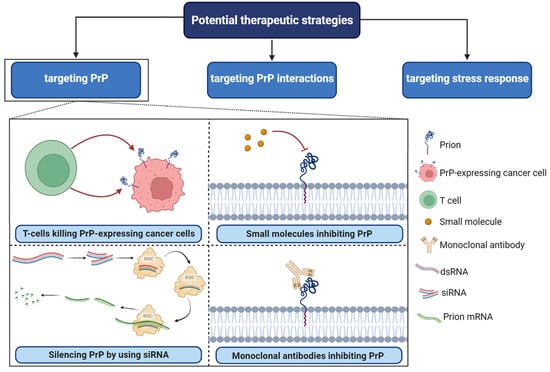
PrPC plays a central role as a scaffold protein by forming multiprotein complexes with receptors or extracellular molecules. These interactions may contribute to the activation of downstream signal pathways that control numerous biological functions, including cancer stem cell self-renewal, the central entity of tumor maintenance and dissemination [30][78]. One potential strategy for targeting PrPC in cancer is to disrupt its interactions with other molecules known to be involved in cancer progression [75]. For example, PrPC has been shown to interact with several cell surface receptors, including integrins and laminin receptors. Importantly, these proteins have been reported to play important roles in cancer cell adhesion, migration, and invasion [69]. Inhibiting these interactions could potentially prevent cancer cells from spreading and invading surrounding tissues. In breast cancer, PrPC interaction with P-gp was associated with drug resistance, higher aggressiveness, invasion, and migration. PrPC also interfered in neo-adjuvant chemotherapy response in this cancer [78].Figure 4. Potential therapeutic strategies in PrPC-associated cancer. RISC; RNA-induced silencing complex, SiRNA; small interfering RNA, dsRNA; double-stranded RNA. Several small molecules, including Quinacrine, Chlorpromazine, Amphotericin B, Pentosan polysulfate, and Suramin, have been identified as potential inhibitors of PrPC.
References
- Bendheim, P.E.; Brown, H.R.; Rudelli, R.D.; Scala, L.J.; Goller, N.L.; Wen, G.Y.; Kascsak, R.J.; Cashman, N.R.; Bolton, D.C. Nearly ubiquitous tissue distribution of the scrapie agent precursor protein. Neurology 1992, 42, 149–156.
- Bonnon, C.; Wendeler, M.W.; Paccaud, J.-P.; Hauri, H.-P. Selective export of human GPI-anchored proteins from the endoplasmic reticulum. J. Cell Sci. 2010, 123 Pt 10, 1705–1715.
- Tanaka, S.; Maeda, Y.; Tashima, Y.; Kinoshita, T. Inositol deacylation of glycosylphosphatidylinositol-anchored proteins is mediated by mammalian PGAP1 and yeast Bst1p. J. Biol. Chem. 2004, 279, 14256–14263.
- Campana, V.; Sarnataro, D.; Zurzolo, C. The highways and byways of prion protein trafficking. Trends Cell Biol. 2005, 15, 102–111.
- Sarnataro, D.; Campana, V.; Paladino, S.; Stornaiuolo, M.; Nitsch, L.; Zurzolo, C. PrP(C) association with lipid rafts in the early secretory pathway stabilizes its cellular conformation. Mol. Biol. Cell 2004, 15, 4031–4042.
- Burns, C.S.; Aronoff-Spencer, E.; Dunham, C.M.; Lario, P.; Avdievich, N.I.; Antholine, W.E.; Olmstead, M.M.; Vrielink, A.; Gerfen, G.J.; Peisach, J.; et al. Molecular features of the copper binding sites in the octarepeat domain of the prion protein. Biochemistry 2002, 41, 3991–4001.
- Harris, D.A.; Lele, P.; Snider, W.D. Localization of the mRNA for a chicken prion protein by in situ hybridization. Proc. Natl. Acad. Sci. USA 1993, 90, 4309–4313.
- Manson, J.; West, J.D.; Thomson, V.; McBride, P.; Kaufman, M.H.; Hope, J. The prion protein gene: A role in mouse embryogenesis? Development 1992, 115, 117–122.
- Brown, D.R.; Schmidt, B.; Groschup, M.H.; Kretzschmar, H.A. Prion protein expression in muscle cells and toxicity of a prion protein fragment. Eur. J. Cell Biol. 1998, 75, 29–37.
- Li, R.; Liu, D.; Zanusso, G.; Liu, T.; Fayen, J.D.; Huang, J.-H.; Petersen, R.B.; Gambetti, P.; Sy, M.-S. The expression and potential function of cellular prion protein in human lymphocytes. Cell. Immunol. 2001, 207, 49–58.
- Pammer, J.; Cross, H.S.; Frobert, Y.; Tschachler, E.; Oberhuber, G. The pattern of prion-related protein expression in the gastrointestinal tract. Virchows Arch. 2000, 436, 466–472.
- Pammer, J.; Weninger, W.; Tschachler, E. Human keratinocytes express cellular prion-related protein in vitro and during inflammatory skin diseases. Am. J. Pathol. 1998, 153, 1353–1358.
- Liemann, S.; Glockshuber, R. Transmissible spongiform encephalopathies. Biochem. Biophys. Res. Commun. 1998, 250, 187–193.
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144.
- Jeffrey, M.; Gonzalez, L. Pathology and pathogenesis of bovine spongiform encephalopathy and scrapie. Curr. Top Microbiol. Immunol. 2004, 284, 65–97.
- Bregman, N.; Shiner, T.; Kavé, G.; Alcalay, R.; Gana-Weisz, M.; Goldstein, O.; Glinka, T.; Aizenstein, O.; Ben Bashat, D.; Alcalay, Y.; et al. Correction: The natural history study of preclinical genetic Creutzfeldt-Jakob Disease (CJD): A prospective longitudinal study protocol. BMC Neurol. 2023, 23, 229.
- Li, B.; Wang, S.; Han, S.; Hu, N.; Shang, X. Case report: Creutzfeldt-Jakob disease: A case that initiated with the onset of obsessive-compulsive state. Front. Neurol. 2023, 14, 1227566.
- Khan, Z.; Bollu, P.C. Fatal Familial Insomnia. In StatPearls; StatPearls: Treasure Island, FL, USA, 2023.
- Cavallaro, U.; Christofori, G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat. Rev. Cancer 2004, 4, 118–132.
- Du, J.; Pan, Y.; Shi, Y.; Guo, C.; Jin, X.; Sun, L.; Liu, N.; Qiao, T.; Fan, D. Overexpression and significance of prion protein in gastric cancer and multidrug-resistant gastric carcinoma cell line SGC7901/ADR. Int. J. Cancer 2005, 113, 213–220.
- Graner, E.; Mercadante, A.F.; Zanata, S.M.; Forlenza, O.V.; Cabral, A.L.; Veiga, S.S.; Juliano, M.A.; Roesler, R.; Walz, R.; Minetti, A.; et al. Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res. Mol. Brain Res. 2000, 76, 85–92.
- Pan, T.; Wong, B.-S.; Liu, T.; Li, R.; Petersen, R.B.; Sy, M.-S. Cell-surface prion protein interacts with glycosaminoglycans. Biochem. J. 2002, 368 Pt 1, 81–90.
- Pan, Y.; Zhao, L.; Liang, J.; Liu, J.; Shi, Y.; Liu, N.; Zhang, G.; Jin, H.; Gao, J.; Xie, H.; et al. Cellular prion protein promotes invasion and metastasis of gastric cancer. FASEB J. 2006, 20, 1886–1888.
- Rieger, R.; Edenhofer, F.; Lasmézas, C.I.; Weiss, S. The human 37-kDa laminin receptor precursor interacts with the prion protein in eukaryotic cells. Nat. Med. 1997, 3, 1383–1388.
- Satoh, J.-I.; Kuroda, Y.; Katamine, S. Gene expression profile in prion protein-deficient fibroblasts in culture. Am. J. Pathol. 2000, 157, 59–68.
- Schmitt-Ulms, G.; Legname, G.; Baldwin, M.A.; Ball, H.L.; Bradon, N.; Bosque, P.J.; Crossin, K.L.; Edelman, G.M.; DeArmond, S.J.; Cohen, F.E.; et al. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 2001, 314, 1209–1225.
- Cheng, Q.; Zheng, H.; Li, M.; Wang, H.; Guo, X.; Zheng, Z.; Chen, C.; Liu, J.; Zhan, T.; Li, Z.; et al. LGR4 cooperates with PrPc to endow the stemness of colorectal cancer stem cells contributing to tumorigenesis and liver metastasis. Cancer Lett. 2022, 540, 215725.
- Kim, M.-J.; Cho, Y.-A.; Kim, E.; Choe, J.-Y.; Park, J.-W.; Lee, J.; Lee, J.-W.; Moon, S.-H.; Kim, Y.-S.; Kim, S.-E.; et al. Cellular Prion Protein Is Closely Associated with Early Recurrence and Poor Survival in Patients with Hepatocellular Carcinoma. Diagnostics 2022, 12, 1635.
- Ding, M.; Chen, Y.; Lang, Y.; Cui, L. The Role of Cellular Prion Protein in Cancer Biology: A Potential Therapeutic Target. Front. Oncol. 2021, 11, 742949.
- Yousaf, S.; Ahmad, M.; Wu, S.; Zia, M.A.; Ahmed, I.; Iqbal, H.M.; Liu, Q.; Rehman, S.U. Cellular Prion Protein Role in Cancer Biology: Is It A Potential Therapeutic Target? Biomedicines 2022, 10, 2833.
- Blasi, F.; Carmeliet, P. uPAR: A versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol. 2002, 3, 932–943.
- Lee, K.S.; Linden, R.; Brentani, R.R.; Martins, V.R.; Prado, M.A.M. Towards cellular receptors for prions. Rev. Med. Virol. 2003, 13, 399–408.
- Limone, A.; Maggisano, V.; Sarnataro, D.; Bulotta, S. Emerging roles of the cellular prion protein (PrP(C)) and 37/67 kDa laminin receptor (RPSA) interaction in cancer biology. Cell. Mol. Life Sci. 2023, 80, 207.
- Li, C.; Yu, S.; Nakamura, F.; Pentikäinen, O.T.; Singh, N.; Yin, S.; Xin, W.; Sy, M.S. Pro-prion binds filamin A, facilitating its interaction with integrin beta1, and contributes to melanomagenesis. J. Biol. Chem. 2010, 285, 30328–30339.
- Brown, D.R.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.; Strome, R.; Fraser, P.E.; Kruck, T.; von Bohlen, A.; Schulz-Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687.
- Ke, J.; Wu, G.; Zhang, J.; Li, H.; Gao, S.; Shao, M.; Gao, Z.; Sy, M.-S.; Cao, Y.; Yang, X.; et al. Melanoma migration is promoted by prion protein via Akt-hsp27 signaling axis. Biochem. Biophys. Res. Commun. 2020, 523, 375–381.
- Lavoie, J.; Gingras-Breton, G.; Tanguay, R.; Landry, J. Induction of Chinese hamster HSP27 gene expression in mouse cells confers resistance to heat shock. HSP27 stabilization of the microfilament organization. J. Biol. Chem. 1993, 268, 3420–3429.
- Nomura, N.; Nomura, M.; Sugiyama, K.; Hamada, J.-I. Phorbol 12-myristate 13-acetate (PMA)-induced migration of glioblastoma cells is mediated via p38MAPK/Hsp27 pathway. Biochem. Pharmacol. 2007, 74, 690–701.
- Clerk, A.; Michael, A.; Sugden, P.H. Stimulation of multiple mitogen-activated protein kinase sub-families by oxidative stress and phosphorylation of the small heat shock protein, HSP25/27, in neonatal ventricular myocytes. Biochem. J. 1998, 333 Pt 3, 581–589.
- McLaughlin, M.M.; Kumar, S.; McDonnell, P.C.; Van Horn, S.; Lee, J.C.; Livi, G.P.; Young, P.R. Identification of mitogen-activated protein (MAP) kinase-activated protein kinase-3, a novel substrate of CSBP p38 MAP kinase. J. Biol. Chem. 1996, 271, 8488–8492.
- New, L.; Jiang, Y.; Zhao, M.; Liu, K.; Zhu, W.; Flood, L.J.; Kato, Y.; Parry, G.C.; Han, J. PRAK, a novel protein kinase regulated by the p38 MAP kinase. EMBO J. 1998, 17, 3372–3384.
- Rouse, J.; Cohen, P.; Trigon, S.; Morange, M.; Alonso-Llamazares, A.; Zamanillo, D.; Hunt, T.; Nebreda, A.R. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell 1994, 78, 1027–1037.
- Santell, L.; Bartfeld, N.S.; Levin, E.G. Identification of a protein transiently phosphorylated by activators of endothelial cell function as the heat-shock protein HSP27. A possible role for protein kinase C. Biochem. J. 1992, 284 Pt 3, 705–710.
- Feng, Y.; Walsh, C.A. The many faces of filamin: A versatile molecular scaffold for cell motility and signalling. Nat. Cell Biol. 2004, 6, 1034–1038.
- Stossel, T.P.; Condeelis, J.; Cooley, L.; Hartwig, J.H.; Noegel, A.; Schleicher, M.; Shapiro, S.S. Filamins as integrators of cell mechanics and signalling. Nat. Rev. Mol. Cell Biol. 2001, 2, 138–145.
- Morelli, J.G.; Yohn, J.J.; Zekman, T.; Norris, D.A. Melanocyte movement in vitro: Role of matrix proteins and integrin receptors. J. Investig. Dermatol. 1993, 101, 605–608.
- Baxter, E.; Windloch, K.; Gannon, F.; Lee, J.S. Epigenetic regulation in cancer progression. Cell Biosci. 2014, 4, 45.
- Kang, T.H.; Noh, K.H.; Kim, J.H.; Bae, H.C.; Lin, K.Y.; Monie, A.; Pai, S.I.; Hung, C.-F.; Wu, T.-C.; Kim, T.W. Ectopic expression of X-linked lymphocyte-regulated protein pM1 renders tumor cells resistant to antitumor immunity. Cancer Res. 2010, 70, 3062–3070.
- Gonzalez-Malerva, L.; Park, J.; Zou, L.; Hu, Y.; Moradpour, Z.; Pearlberg, J.; Sawyer, J.; Stevens, H.; Harlow, E.; LaBaer, J. High-throughput ectopic expression screen for tamoxifen resistance identifies an atypical kinase that blocks autophagy. Proc. Natl. Acad. Sci. USA 2011, 108, 2058–2063.
- Meijer, D.; van Agthoven, T.; Bosma, P.T.; Nooter, K.; Dorssers, L.C. Functional screen for genes responsible for tamoxifen resistance in human breast cancer cells. Mol. Cancer Res. 2006, 4, 379–386.
- Kandasamy, K.; Srinivasula, S.M.; Alnemri, E.S.; Thompson, C.B.; Korsmeyer, S.J.; Bryant, J.L.; Srivastava, R.K. Involvement of proapoptotic molecules Bax and Bak in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced mitochondrial disruption and apoptosis: Differential regulation of cytochrome c and Smac/DIABLO release. Cancer Res. 2003, 63, 1712–1721.
- Keane, M.M.; Ettenberg, S.A.; Nau, M.M.; Russell, E.K.; Lipkowitz, S. Chemotherapy augments TRAIL-induced apoptosis in breast cell lines. Cancer Res. 1999, 59, 734–741.
- Kim, E.J.; Suliman, A.; Lam, A.; Srivastava, R.K. Failure of Bcl-2 to block mitochondrial dysfunction during TRAIL-induced apoptosis. Tumor necrosis-related apoptosis-inducing ligand. Int. J. Oncol. 2001, 18, 187–194.
- Simstein, R.; Burow, M.; Parker, A.; Weldon, C.; Beckman, B. Apoptosis, chemoresistance, and breast cancer: Insights from the MCF-7 cell model system. Exp. Biol. Med. 2003, 228, 995–1003.
- Sprick, M.R.; Rieser, E.; Stahl, H.; Grosse-Wilde, A.; Weigand, M.A.; Walczak, H. Caspase-10 is recruited to and activated at the native TRAIL and CD95 death-inducing signalling complexes in a FADD-dependent manner but can not functionally substitute caspase-8. EMBO J. 2002, 21, 4520–4530.
- Srivastava, R.K. Intracellular mechanisms of TRAIL and its role in cancer therapy. Mol. Cell Biol. Res. Commun. 2000, 4, 67–75.
- Meslin, F.; Hamai, A.; Gao, P.; Jalil, A.; Cahuzac, N.; Chouaib, S.; Mehrpour, M. Silencing of prion protein sensitizes breast adriamycin-resistant carcinoma cells to TRAIL-mediated cell death. Cancer Res. 2007, 67, 10910–10919.
- Gil, M.; Kim, Y.K.; Kim, K.-E.; Kim, W.; Park, C.-S.; Lee, K.J. Cellular prion protein regulates invasion and migration of breast cancer cells through MMP-9 activity. Biochem. Biophys. Res. Commun. 2016, 470, 213–219.
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434.
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314.
- Li, Q.-Q.; Sun, Y.-P.; Ruan, C.-P.; Xu, X.-Y.; Ge, J.-H.; He, J.; Xu, Z.-D.; Wang, Q.; Gao, W.-C. Cellular prion protein promotes glucose uptake through the Fyn-HIF-2α-Glut1 pathway to support colorectal cancer cell survival. Cancer Sci. 2011, 102, 400–406.
- De Wit, M.; Jimenez, C.R.; Carvalho, B.; Belien, J.A.; Delis-van Diemen, P.M.; Mongera, S.; Piersma, S.R.; Vikas, M.; Navani, S.; Pontén, F.; et al. Cell surface proteomics identifies glucose transporter type 1 and prion protein as candidate biomarkers for colorectal adenoma-to-carcinoma progression. Gut 2012, 61, 855–864.
- Wang, Q.; Qian, J.; Wang, F.; Ma, Z. Cellular prion protein accelerates colorectal cancer metastasis via the Fyn-SP1-SATB1 axis. Oncol. Rep. 2012, 28, 2029–2034.
- Han, H.; Bearss, D.J.; Browne, L.W.; Calaluce, R.; Nagle, R.B.; Von Hoff, D.D. Identification of differentially expressed genes in pancreatic cancer cells using cDNA microarray. Cancer Res. 2002, 62, 2890–2896.
- Lee, J.H.; Yun, C.W.; Lee, S.H. Cellular Prion Protein Enhances Drug Resistance of Colorectal Cancer Cells via Regulation of a Survival Signal Pathway. Biomol. Ther. 2018, 26, 313–321.
- Li, C.; Yu, S.; Nakamura, F.; Yin, S.; Xu, J.; Petrolla, A.A.; Singh, N.; Tartakoff, A.; Abbott, D.W.; Xin, W.; et al. Binding of pro-prion to filamin A disrupts cytoskeleton and correlates with poor prognosis in pancreatic cancer. J. Clin. Investig. 2009, 119, 2725–2736.
- Sy, M.-S.; Altekruse, S.F.; Li, C.; Lynch, C.F.; Goodman, M.T.; Hernandez, B.Y.; Zhou, L.; Saber, M.S.; Hewitt, S.M.; Xin, W. Association of prion protein expression with pancreatic adenocarcinoma survival in the SEER residual tissue repository. Cancer Biomark 2011, 10, 251–258.
- Chieng, C.K.-L.; Say, Y.-H. Cellular prion protein contributes to LS 174T colon cancer cell carcinogenesis by increasing invasiveness and resistance against doxorubicin-induced apoptosis. Tumour. Biol. 2015, 36, 8107–8120.
- Sauer, H.; Dagdanova, A.; Hescheler, J.; Wartenberg, M. Redox-regulation of intrinsic prion expression in multicellular prostate tumor spheroids. Free. Radic. Biol. Med. 1999, 27, 1276–1283.
- Sollazzo, V.; Galasso, M.; Volinia, S.; Carinci, F. Prion proteins (PRNP and PRND) are over-expressed in osteosarcoma. J. Orthop. Res. 2012, 30, 1004–1012.
- Thellung, S.; Corsaro, A.; Bosio, A.; Zambito, M.; Barbieri, F.; Mazzanti, M.; Florio, T. Emerging Role of Cellular Prion Protein in the Maintenance and Expansion of Glioma Stem Cells. Cells 2019, 8, 1458.
- Yang, C.C.; Chuang, F.C.; Chang, C.L.; Huang, C.R.; Chen, H.H.; Yip, H.K.; Chen, Y.T. Melatonin-Assisted Cisplatin Suppresses Urinary Bladder Cancer Cell Proliferation and Growth through Inhibiting PrP(C)-Regulated Cell Stress and Cell Proliferation Signaling. Int. J. Mol. Sci. 2023, 24, 3353.
- Ameyar-Zazoua, M.; Larochette, N.; Dorothée, G.; Daugas, E.; Haddada, H.; Gouloumet, V.; Métivier, D.; Stancou, R.; Mami-Chouaib, F.; Kroemer, G.; et al. Wild-type p53 induced sensitization of mutant p53 TNF-resistant cells: Role of caspase-8 and mitochondria. Cancer Gene Ther. 2002, 9, 219–227.
- Lenzi, P.; Busceti, C.L.; Lazzeri, G.; Ferese, R.; Biagioni, F.; Salvetti, A.; Pompili, E.; De Franchis, V.; Puglisi-Allegra, S.; Frati, A.; et al. Autophagy Activation Associates with Suppression of Prion Protein and Improved Mitochondrial Status in Glioblastoma Cells. Cells 2023, 12, 221.
- Mouillet-Richard, S.; Martin-Lannerée, S.; Le Corre, D.; Hirsch, T.Z.; Ghazi, A.; Sroussi, M.; Pilati, C.; de Reyniès, A.; Djouadi, F.; Vodovar, N.; et al. A proof of concept for targeting the PrP(C)—Amyloid beta peptide interaction in basal prostate cancer and mesenchymal colon cancer. Oncogene 2022, 41, 4397–4404.
- Yu, G.; Jiang, L.; Xu, Y.; Guo, H.; Liu, H.; Zhang, Y.; Yang, H.; Yuan, C.; Ma, J. Silencing prion protein in MDA-MB-435 breast cancer cells leads to pleiotropic cellular responses to cytotoxic stimuli. PLoS ONE 2012, 7, e48146.
- Kouadri, A.; El Khatib, M.; Cormenier, J.; Chauvet, S.; Zeinyeh, W.; El Khoury, M.; Macari, L.; Richaud, P.R.; Coraux, C.; Michaud-Soret, I.; et al. Involvement of the Prion Protein in the Protection of the Human Bronchial Epithelial Barrier Against Oxidative Stress. Antioxid Redox Signal. 2019, 31, 59–74.
- Linden, R.; Martins, V.R.; Prado, M.A.M.; Cammarota, M.; Izquierdo, I.; Brentani, R.R. Physiology of the prion protein. Physiol. Rev. 2008, 88, 673–728.
More