Alzheimer’s Disease (AD), the most common type of dementia, is known as a neurodegenerative disease caused by the accumulation of amyloid beta (Aβ) peptides and tau protein hyperphosphorylation resulting in the formation of neurofibrillary tangles,. activation of inflammasomes, sluggish autophagy, and neuronal loss. Several of these hallmarks are linked to alteration in the gut microbiome, also known as gut dysbiosis. SelectiveCatechins are a group of bioflavonoids can target gut microbiome to inhibit inflammasomes and resume autophagy to stop AD pathogenesis. Two bioflavonoids, specifically epigallothat can be extracted from tea, and this group includes epigallocatechin (EGC), epicatechin-3- gallate (EGCG) and genistein (GS), appear to be a new paradigm of treatment for maintaining healthy gut microbiome in AD via modulating crucial AD signaling pathwaysCG), epicatechin (EC), and the most abundant compound EGCG.
- Alzheimer’s disease (AD)
- autophagy
- bioflavonoids
- epigallocatechin-3-gallate (EGCG)
- genistein (GS)
- gut microbiome
1. Introduction
2. BPrescriptioflavonoids as Novel on Therapeutic Options for AD
The cholinere are gic hypothesis states that the onset of AD progresses due to the decrease in acetylcholine (ACh) synthesis [10]. Hence, this therapeutic strategy intends to inhibit the activity of arying potecetylcholinesterase enzyme (AChE), which otherwise degrades ACh, to increase the cholinergic signaling in the brain. By deterring the degradation of ACh at the synapses, the cholinergic receptors stay activated [5]. To inhibit tial categories for thehe AChE, varying AChE inhibitors have been created, such as Physostigmine, Tacrine, Donepezil, Rivastigmine, Galantamine, and Metrifonate. Among these inhibitors, only four drugs as potential therapeutics, including Donepezil (AChE inhibitor), Galantamine (AChE inhibitor), Rivastigmine (reverse inhibitor of both AChE and butyrylcholinesterase or BChE), and Memantine are currently available in the market for use in the AD patients (Table 1). However, all these drugs have side effects, which increase with the increasing dosage administered [10].| AChE Inhibitor | Dosage (mg/d) |
Outcomes and Side Effects | Availability for Clinical Use | Reference |
|---|---|---|---|---|
| Tacrine | 80–160 | Nausea and abnormal liver functionality | No | [10] |
| Physostigmine | 36 | Nausea, diarrhea, and dizziness | No | [11] |
| Rivastigmine | 6–12 | Nausea, vomiting, and diarrhea | Yes | [11] |
| Galantamine * | 20–50 | Nausea, vomiting, and diarrhea | Yes | [11] |
| Donepezil | 10 | Higher cognitive improvements and reduced inflammatory cytokines as well as oxidative stress | Yes | [12] |
| 5 | Reduced inflammatory cytokines and oxidative stress | |||
| Metrifonate | NA | Bradycardia, rhinitis, abdominal pain, neuromuscular dysfunction, and respiratory failure | No | [13] |
3. Bioflavonoids as Novel Therapeutic Option for AD
As mentioned above, there are varying potential categories for therapeutic values of AChE inhibitors in the treatment of AD. However, natural compounds derived from plants are currently gaining wide attention for the treatment of AD and other neurodegenerative diseases in vitro and in vivo. Polyphenols are known to have enormous potential to regulate diversity as well as the composition of gut microbiota, which is associated with neurological health. Studies have showcased that polyphenols can reduce the neurological deficits that are caused due to neuroinflammation [10][11][12][14,15,16]. Bioflavonoids are a group of natural polyphenolic compounds that are derived from fruits and vegetables. A few examples of fruits and vegetables would include apples, onions, mulberries, and bilberries. Bioflavonoids are popularly consumed via tea, beer, and wine [13][17]. This subclass of polyphenols of biological origin is implicated in anti-apoptotic and pro-survival signaling pathways and decreasing the pathological effects of AD [14][15][18,19]. Among the 5000 types of flavonoids, which are mostly found in plants, there are six main types of flavonoids such as flavonols, flavones, flavan-3-ols, flavanones, anthocyanidins, and isoflavones [20,21]. Bioflavonoids, which are exclusively derived from biological origins (mainly plants), are also well known for showcasing anti-inflammatory, anti-viral, anti-apoptotic, anti-platelet, and anti-tumoral properties [13][18][19][17,22,23]. Catechins are a group of bioflavonoids that can be extracted from tea, and this group includes epigallocatechin (EGC), epicatechin gallate (ECG), epicatechin (EC), and the most abundant compound EGCG [19][23]. As shown in Figure 1, the chemical structure of EGCG contains A, B, C, and D rings produced from the esterification of EGC with gallic acid [20][24]. Both the A and C rings have a phenyl group at C2 and a gallate group at C3 positions. The B and D rings of EGCG contain 3,4,5-trihydroxy groups, which have the potential for proteasome activity in vitro [20][24]. On the aromatic B ring, catechins have di- or tri-hydroxyl groups along with meta-5,7-dihydroxyl groups on the A ring [21][25]. The presence of phenolic groups in these compounds increases their antioxidant properties. The structure of flavonoids is important in creating a novel therapeutic drug for the treatment of AD.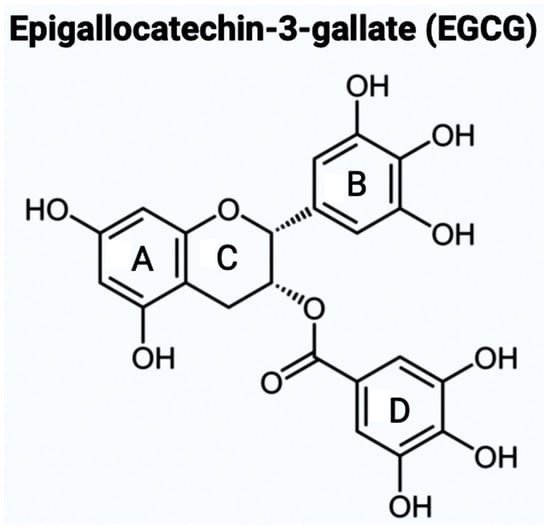
34. An Overview of the Gut Microbiota
The gut microbiota in every individual is unique, and thus, it is determined through environmental factors rather than being a genetically inheritable trait. In the gut microbiota, the staggering microbial diversity and colonization result in varying complex interactions, diseases, and immune responses. Depending on the area of the gastrointestinal tract being examined, the density and diversity of the gut bacteria fluctuate due to the difference in the local conditions [25][29]. A proper understanding of the function of microbiota in the gut is crucial for the development of successful therapeutics to target neurological disorders. There are diverses inclu groups of gut members (microbiota, microbial structural elements, microbial metabolites, and internal/external structural elements) that morph into the concept of the microbiome [28]. Gut microbiota contributes to all the living organisms present in the microbiome, such as bacteria, archaea, fungi, protists, and algae. In comparing Ason, microbial structural elements include a variety of components such as proteins, lipids, polysaccharides, nucleic acids, and other genetic elements. Signaling molecules, toxins, and organic molecules generate a group of microbial metabolites. Additionally, environmental conditions also affect the ecological niche of the human body, resulting in the variability of the microbiome in people [28]. In the human body, microbial cells are as abundantly found as somatic cells. At the current estimation, there are 500–1000 or more species of bacteria existing in the human body [28]. In gut microbiota, the existing bacteria belong to two different phyla called Bacteroidetes and Firmicutes, and both phyla consist of more than 200 genera [29,30]. The extent of microbial colonization for versatile roles in the human body can be appreciated through an approximate estimate of 2,000,000 bacterial genes in the gut compared to about 20,000 human genes [28]. However, due to the scale and complexity of the microbial diversity present, the factors and influences affecting the conditions of the microbiome, along with the resulting interactions with the immune, endocrine, and nervous systems, are being heavily researched in many disease pathologies including AD.3.1. Activity of Gut Microbiota in the Human Body
4.1. Activity of Gut Microbiota in the Human Body
The intestinal bacteria produce short-chain fatty acids (SCFAs) through the fermentation of non-digestible carbohydrates (NDC) and dietary fiber, and these SCFAs are formate, acetate, propionate, and butyrate, with the presence of acetate being three times higher [26][27][28][30,31,32]. Specifically, an increased presence of formate has been linked to the possibility of higher inflammation [28][32]. In a study of the introduction of butyrate to isolated germ-free colonocytes, the rate of oxidative phosphorylation increased while autophagy decreased [27][31]. For fermentation, the source of the carbohydrates used is the ones that were not digested or absorbed in the small intestine [28][32]. The functions of SCFAs include affecting cellular processes such as gene expression, differentiation, proliferation, and apoptosis [27][31]. The SCFAs, which are produced from NDC and dietary fiber, also regulate the permeability of the gut and blood-brain barriers [29][33]. In the CNS, SCFAs play a key role during the production of neural progenitor cells (NPCs) that produce neuronal and glial cell types [30][31][34,35]. According to a study conducted recently, the increased concentration of SCFAs positively affected the expression of genes involved in the proliferation of NPCs [32][36]. Free fatty acids (FFAs), which are the products of the metabolic pathways, are known to function as signaling molecules via interaction with free fatty acid receptors (FFARs) that form a family of G protein-coupled receptors (GPCRs). As the largest group of transmembrane proteins, GPCRs are currently known to be the most successful drug targets. There are a few mechanisms that are regulated by SCFAs to increase the production of NPCs. The first mechanism involves specific FFARs (i.e., GPCRs), most notably FFAR2 (GPR43) and FFAR3 (GPR41), being upregulated due to increased exposure to SCFAs. In the second mechanism, SCFAs regulate the physiological pH that modulates neurodevelopmental effects along with anti-apoptotic effects [36]. This [32]showcases the importance of further exploring the influence of the SCFAs in the gut microbiome regarding diseases, especially neurologically related. SCFAs are produced in the intestinal tract due to the metabolic activities of the diverse and thriving bacteria population there. The concentrations of SCFAs vary based on the location in the gut [31]. In the proximal colon, 70 to 140 mM SCFAs are present, and in the distal colon, they are about 20 to 70 mM with increased production of acetate. Additionally, SCFAs have established themselves in the oral cavity as well, with varying concentrations of the different fatty acids (6 to 38 mM acetate, 1 to 13 mM propionate, and 0 to 5 mM butyrate). In the lower female genital tract, the acetate concentration can reach up to 120 mM, which is directly influenced by infection or inflammation [31]. Regarding inflammation, SCFAs modulate the production of cytokines, which are crucial to control inflammation [30]. Besides cytokines, SCFAs also regulate other immune cells, including macrophages, neutrophils, and dendritic cells. According to studies, Faecalibacterium prausnitzii (also called F. prausnitzii, a strictly anaerobic bacterium) has an inflammatory protein that can inhibit NF-κB family of transcription factors (present in the intestinal epithelial cells of the animals). This transcription factor family begins the transcription of target genes by binding to a specific DNA element, κB enhancer. After one of the five transcription factors belonging to the family activates the κB enhancer, the phosphorylated inhibitor of NF-κB or IκB is degraded using a proteasome. This results in the NF-κB being freed from the cytoplasm and moved to the nucleus to activate pro-inflammatory genes to heal tissue damage through cytokine production [30,37]. The importance of NF-κB pathway in targeting pro-inflammatory genes can lead to an overactivation of this pathway, resulting in higher levels of inflammation [38]. To suppress the overproduction of inflammatory cytokines, the gut microbiota can inhibit the NF-κB pathway using the microbe-derived factors [38].3.2. Onset Factors in the Microbiota for Dysbiosis
4.2. Onset Factors in the Microbiota for Dysbiosis
Dysbiosis occurs when the normal state of the gut microbiota is unbalanced due to varying factors. One of the main examples is when the anti-inflammatory cytokines and the pro-inflammatory cytokines produced by the microbes are not balanced, then dysbiosis takes place. This imbalancere are t can result in conditions including inflammatory bowel disease (IBD), irritable bowel syndrome (IBS), diabetes, obesity, cancer, cardiovascular problems, and many CNS disorders [39]. Both IBD and IBS aree t incredibly challenging conditions. There are three types of dysbiosis: type 1 indicates a decrease of beneficial bacteria, type 2 shows an increase of pathogenic bacteria, and type 3 states a decrease in overall bacterial diversity [34][40]. The number of factors that can directly affect dysbiosis are many such as diet, birthing conditions (e.g., mode of birth, antibiotic exposure, and hygiene), chemical exposure, psychological and environmental stimuli (e.g., pathogens, sleep deprivation, circadian rhythm dysfunction, toxins, and noise), temperature, and intestinal infection. Diet is one of the crucial regulators of the gut microbiota [26][30]. In studies comparing a ‘Western diet’ (high animal protein, high in sugar and saturated fats) and an ‘agrarian diet’ (low animal protein, low levels of saturated fat and simple sugars), the results displayed the ‘Western diet’ leading to dysbiosis and lower levels of SCFAs [35][41]. On the other hand, the ‘agrarian diet’ results in more production of SCFAs and higher gut bacteria diversity, which helps limit the growth of potentially pathogenic bacteria that otherwise lead to diseases such as IBD [35][41]. The reason why the lower animal protein levels in an ‘agrarian diet’ help with the gut microbiota is due to the side effects of protein and amino acid fermentation [36][42]. When more protein is consumed, the gut must shift to increase the pH to break down the proteins that result in the production of compounds, including hydrogen sulfide, reactive oxygen species, and ammonia, which are unhealthy for the gut [42,43]. An increased intake of Vitamin D can help inhibit inflammatory responses along with modulating the state of the gut microbiota. When mice lack Vitamin D, the intestinal epithelial barrier is not protected, resulting in the growth of pathogenic bacteria in the gut and the initiation of inflammation due to dysbiosis [41]. Apart from the influence of diet on dysbiosis, environmental factors are also implicated in this process, taking place in the microbiota. Beginning from birth, a person’s gut flora is affected based on whether they were born vaginally or via a c-section [44]. Studies have showcased a relationship between how likely a child is to develop obesity/diabetes and whether they were born vaginally [45]. In the case of children who were born vaginally, they were reported to have been exposed to the mother’s beneficial bacteria present in the birth canal and rectum. On the other hand, c-section babies were stated to be at a higher risk of developing diabetes because they were only exposed to pathogenic bacteria [45]. The factors that cause dysbiosis in the gut microbiota are diverse, so exploring the gut flora in relation to diseases is [36][37]crucial.3.3. Gut-Brain Axis (GBA) and Gut Dysbiosis
4.3. Gut-Brain Axis (GBA) and Dysbiosis
The relationship between the gut microbiota and the brain is called the gGut-brain aBrain Axis (GBA), and this two-way communication is built using immune, circulatory, and neural pathways (Figure 3) [30][34]. GBA connects the CNS (comprised of brain and spinal cord), autonomic nervous system (ANS), enteric nervous system (ENS), and hypothalamic pituitary adrenal (HPA) axis [38][46]. Particularly, the function of the HPA axis includes regulating the adaptive responses from the body to any stressors needed [38][39][46,47]. An increase in the occurrence of inflammatory cytokines such as interleukin-1 beta (IL-1β), IL-6, and tumor necrosis factor alpha (TNF-α) through the production of corticotropin-releasing factor (CRF) and adrenocorticotropic hormone (ACTH) is an example of environmental stress that can activate the HPA axis. The bidirectional communication line results in the regulation of intestinal functional effector cells (immune cells, epithelial cells, enteric neurons, smooth muscle cells, interstitial cells of Cajal, and enterochromaffin cells) [38][46]. Unsurprisingly, the gut microbiota has been implicated in affecting the bidirectional communication between the gut and the brain [38][46]. The microbes present in the gut produce metabolites such as SCFAs, gamma-aminobutyric acid (GABA), tryptophan, serotonin, catecholamines, metabolites of bile acids and neurotransmitters, and cytokines that can signal to the receptors present in the gut [48]. [40]The sequence of the events leading up to dysbiosis in the gut has not yet been properly established and can be caused by varying stresses. The dysbiosis of the gut microflora causes an increase in the gut and blood-brain barrier permeability, production of bacterial amyloids, and formation of lipopolysaccharides (LPS) leading up to the deposition of amyloid fibrils in the brain, resulting in the pathogenesis (neuroinflammation, cognitive decline) of neurological disorders such as AD and stroke [41][49]. There still has not been a complete understanding of the pathways involved in the GBA bidirectional communication line.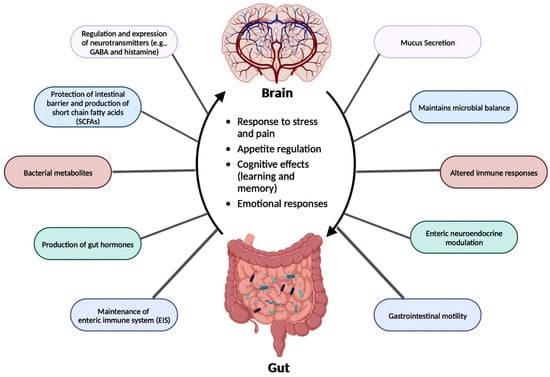
3.4. GBA and AD
4.4. GBA and AD
The dysbiosis in gut microbiota directly affects the GBA, which is linked to AD clinical symptoms such as Aβ plaque deposition, cognitive decline, and memory loss (Table 12). To explore the change in the microbiota and the Aβ plaque deposition in the brain, studies created AD animal models comparing the microorganisms and SCFAs in fecal samples between AD mice and wild-type animals [42][51]. The AD mice displayed lower levels of SCFAs, which had the potential to alter multiple metabolic pathways along with increasing the deposition of Aβ plaques [42][51]. Another study for analyzing the effect of age compared the double transgenic (TG) mice expressing a chimeric mouse/human amyloid precursor protein (APP) and a mutant human presenilin 1 (PSEN1), both APP/PSEN1 mutations ensured an early-onset of D, and C57BL/6 wild-type (WT) mice, and the results unveiled the potential of targeting the gut microbiota in AD animals [43][52]. The 6-month-old APP/PSEN1 mice, with their gut microflora documented differently from the WT mice, experienced cognitive decline [43][52]. The microbial diversity of the APP/PSEN1 mice deteriorated along with age with increases in the population of bacteria from the Helicobacteraceae and Desulfovibrionaceae families [43][52]. In the Helicobacteraceae family, Helicobacter pylori (H. pylori) participates in causing dysbiosis resulting in gastric disorders such as chronic active gastritis, peptic ulcer disease (PUD), mucosa-associated lymphoid tissue (MALT) lymphoma and gastric carcinoma [44][53]. Interestingly, a recent study revealed inserting Desulfovibrio stains in Caenorhabditis elegans (C. elegans) increased the number of alpha-synuclein aggregates causing Parkinson’s disease (PD), another prominent neurodegenerative disease [54]. The decrease in microbial diversity in the APP/PSEN1 mice highlights the importance of regulating the gut microbiome as a viable therapeutic target for the treatment of AD.| AD Animal Model | Change in Gut Microbiota in AD Mice |
Observed Pathological Symptoms |
Reference |
|---|---|---|---|
| AD model mice (with varying ages) | Decreased microbial diversity and reduced SCFA levels | Amyloid deposition and ultrastructural abnormalities in the intestine, cognitive dysfunction, and signaling pathway alterations | [42][51] |
| APP/PSEN1 mice | Decreased microbial diversity | Cognitive dysfunction | [43][52] |
| APPSWE/PSIΔE9 mice | Varied gut microbial composition | Increased cerebral Aβ pathology | [46][55] |
| APP/PS1 mice | Increased pro-inflammatory bacteria during aging | Autism and inflammatory-related disorders | [47][56] |
| ApoE-/- mice | Porphyromonas gingivalis infection | Neuronal injury | [48][57] |
4
Beyond hypothesizing the link between the gut microbiota and the brain regarding AD pathogenesis, there are recent studies that display a possibility of AD development beginning in the gut before reaching the brain. In the gastric wall of mice, Aβ1–42 oligomers were administered to induce amyloidosis [59]. The amyloid was observed using immunofluorescence staining and in vivo imaging to have traveled from the gut to the brain [59]. Various AD hallmarks were observed in the mice, such as cerebral and vagal beta-amyloidosis. Another remarkable finding was gastric dysfunction, which was usually attributed to PD symptoms [59]. Consequently, the exploration of the origin of AD pathogenesis should be extended beyond focusing on the brain and include the gut microbiota.5. Neuroinflammation in AD
Most neurological conditions, such as autism spectrum disorders (ASD), epilepsy, PD, cerebrovascular diseases, and AD, have aspects of neuroinflammation as part of their pathogenesis. Cytokines, produced by microglia and astrocytes, are the central factors that influence all characteristics of neuroinflammation, ranging from pro-inflammatory and anti-inflammatory processes to neuronal injury [50][60]. In the case of neurological disorders, microglial activation results in the production of pro-inflammatory cytokines (e.g., IL-1, IL-6, and TNF-α), and overproduction of these molecules is toxic to neurons and glial cells [61]. In AD, Aβ forms soluble oligomers and fibrils, which bind to microglia through cell-surface receptors, including the scavenger receptor class a1 (SCARA1), a cluster of differentiation 36 (CD36), CD14, a6β1 integrin, CD47, and Toll-like receptors (TLRs). Hence, the deletion of CD36, TLR4, or TLR6 can decrease Aβ-induced cytokine production [61]. Another source of cytokines is astrocytes, which regulate synaptic transmission through reactive astrogliosis [61]. The activation of astrocytes can happen because of multiple triggers, such as transforming growth factor-beta 1 (TGF-β1), leukemia inhibitory factor (LIF), and ciliary neurotrophic factor [62]. After activation, astrocytes either become neurotoxic A1 producing the pro-inflammatory molecules such as reactive oxygen species (ROS), IL-6, IL-1β, and TNF-α or become neuroprotective A2 along with the protective factors such as vascular endothelial growth factor (VEGF), brain-derived neurotrophic factor (BDNF), and nerve growth factor (NGF) [62]. The exposure of astrocytes to initial Aβ deposits upregulates the expression of Aβ proteases such as neprilysin, insulin-degrading enzyme (IDE), endothelin-converting enzymes (ECE), and angiotensin-converting enzyme (ACE) [60]. In studies exploring animal models of AD, astroglia atrophy was detected in the initial stages. This atrophy, along with dysfunction of Aβ proteases, can cause a decrease in proteolytic clearance of Aβ. Uncontrolled cytokine production negatively impacts the long-term potentiation (LTP) of synaptic transmission, aiding the symptomatic progression of neurodegenerative diseases and increasing intestinal permeability [60,63]. The importance of controlling inflammation in AD could be understood through the example of a clinical study in which AD patients were administered non-steroidal anti-inflammatory drugs (NSAIDs) in the Baltimore longitudinal study, and these patients experienced less cognitive decline compared to the patients who were administered aspirin [64]. Due to the impact of pro-inflammation cytokines produced by both microglia and astrocytes, neuroinflammation is an important target when creating a potential therapeutic drug for AD.4.1. Implications of Gut Microbiota in Neuroinflammation in AD
5.1. Implications of Gut Microbiota in Neuroinflammation in AD
A trigger for inflammation is the structural components of bacteria, including the by-products (e.g., SCFAs, enzymes, metabolites, LPS, cell capsule carbohydrates, and endotoxins) produced during the metabolic processes involved [51][64]. Chronic low-grade inflammation or inflammaging is found to cause tissue damage in most age-related diseases, including AD. One of the causes of inflammaging is dysbiosis in the gut microbiota [52][50]. Studies exploring the inflammation caused by an imbalance in intestinal immunity confirm the involvement of gut microbiota in innate and adaptive immunity, especially in IBD, which eventually can result in PD [28][32]. These studies use sterile-raised germ-free (GF) mice lacking the microorganisms existing in the gastrointestinal (GI) tract, along with mice without pathogens treated with broad-spectrum antibiotics (ABX). The ABX mice represented the innate immune system in which the myeloid cells in the bone marrow were impaired, resulting in a decrease of granulocytes and, thus, a higher likelihood of bacterial infection. In the GF mice, the development of innate lymphoid cells (ILCs) was disabled, leading to antigen receptors not being expressed. Additionally, this lack of expression affects enteric bacterial infections because the production of IL-22 decreases [28][32]. Considering the pathogenesis of PD is closely related to AD, the triggers for extreme inflammation could be shared between the two proteinopathies (aberrant protein aggregate diseases). Colorectal cancer (CRC) is another disease in which dysbiosis in the gut microbiota has a direct association with inflammation. A study analyzing the differences in mucosal samples between patients with CRC adenocarcinoma, tubular adenomas, and intact colon was conducted [65]. The varying bacterial diversity in mucosal samprominenles of participants with tubular adenoma and adenocarcinoma showed signs of dysbiosis [65]. The prominent inflammatory markers generated by the gut microbiota include LPS, SCFAs, bile acids (BAs), C-reactive protein (CRP), and cytokines. The first marker mentioned, LPS, also called endotoxin, is a part of the cell wall of Gram-negative bacteria [54][66]. In a normal gut state, LPS (concentration ranges from 0 to 1.0 ng·mL−1) is prevented by the gut barrier (intestinal epithelial and mucosal layers) from entering systemic circulation and activating epithelial destruction [55][54][63,66]. However, as shown in Figure 4, LPS increased enterocyte membrane TLR-4 expression in animal models of inflammation [55][63].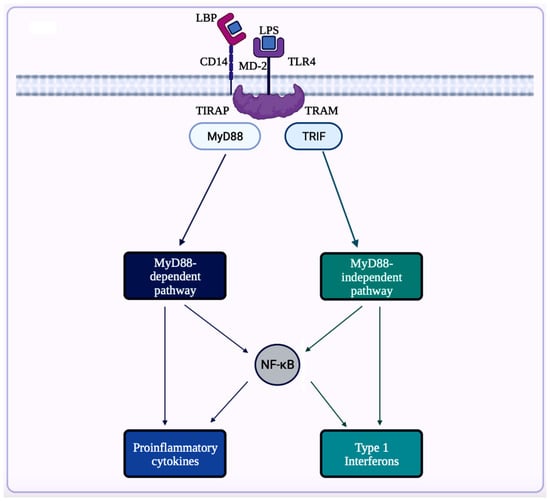
4.2. Inflammasomes in AD
5.2. Inflammasomes in AD
Inflammasome, which is a cytosolic multiprotein complex, is implicated in causing excessive inflammation in various diseases, including autoimmune diseases, cancers, and neurodegenerative diseases [59][62]. In the innate immune system, if adverse stimuli (e.g., pathogens, dead cells) are detected, then inflammasomes are the receptors deployed to activate caspase-1, which contains a caspase recruitment domain (CARD), resulting in inflammation [51][64]. The innate immune response cascade begins with the initiation of germline-encoded pattern recognition receptors (PRRs), transmembrane or cytosolic receptors, by pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) [60][67]. PAMPs are the structural moieties present in microorganisms such as Gram-negative bacterial LPS, bacterial or viral nucleic acids, and bacterial peptides (e.g., flagellin). DAMPs are the endogenous molecules activated due to cellular stress, such as chromatin-associated proteins, heat-shock proteins, uric acid, and extracellular matrix fragments [60][67]. After the PAMPs or the DAMPs activate the cascade, PRRs such as nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs), absent in melanoma-2 (AIM-2)-like receptors (ALRs), and tripartite motif-containing (TRIM) proteins form inflammasomes. There are three diverse types of inflammasomes: NLR-associated inflammasomes, ALR-associated inflammasomes, and the pyrin inflammasome [59][62]. Inflammasomes can be activated through two kinds of inflammasome signaling such as canonical and non-canonical [54][66]. The canonical inflammasome signaling consists of one or more inflammasome sensors, such as apoptosis-associated speck-like protein containing CARD (ASC) and caspase-1. ASC has a bipartite structure with a pyrin domain (PYD) and a CARD, both of which aid ASC in acting as an adaptor molecule. The non-canonical signaling pathway includes the activation of mouse caspase-11 or human caspase-4 and caspase-5 [51][60][64,67]. The NLRP3 (NOD-, leucine-rich repeat- or LRR-, and PYD-containing protein 3) inflammasome is a multimeric protein complex – which is made up of the sensor protein NLRP3, the adaptor protein ASC, and the effector protein pro-caspase-1 – having a role in development of AD [61][62][63][73,74,75]. When the sensor protein NLRP3 is activated, it binds to the PYD of the adaptor protein ASC, resulting in the cleavage of pro-caspase-1 into activate caspase-1 to form the NLRP3 inflammasome (Figure 5) [64][76]. This activates caspase-1, which then activates the inactive pro-inflammatory cytokines pro-IL-1β and pro-IL-18 into their respective mature forms [64][76]. Along with activating cytokines, the activated caspase-1 can also cause pyroptosis, an inflammatory-related programmed cell death [64][76]. Among the family of inflammasomes, NLRP3 is the most extensively studied [63][75].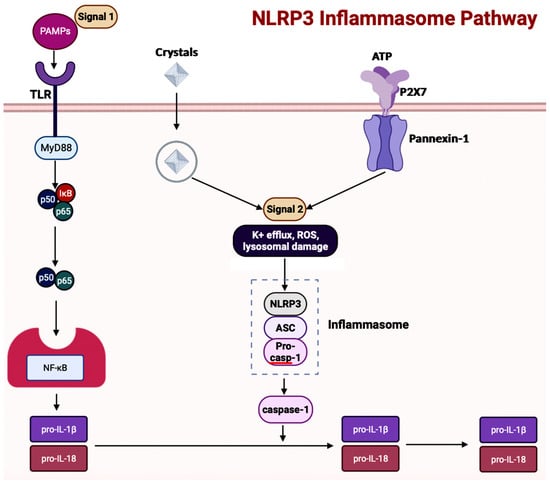
4.3. Inflammasomes and GBA in AD
5.3. Inflammasomes and GBA in AD
Inflammasome activity is influenced by alterations in the gut microbiota and diet [58][72]. In a ketogenic diet or calorie restriction, the NLRP3 inflammasome gets inhibited because the ketone body β-hydroxybutyrate production in the liver increases [62][74]. A study exploring sickness-induced anorexia analyzed the relationship between Salmonella typhimurium and the GBA [69][81]. The S. typhimurium effector, Slrp, was used to inhibit the inflammasome pathway to hinder anorexia [69][81]. In the blood and brain samples collected from patients experiencing cognitive decline, an overexpression of NLRP3 in astrocytes and microglia resulting in central inflammation was witnessed [70][82]. Contrastingly, the same study also analyzed the effects of dysbiosis on the activation of peripheral inflammation, consisting of the innate cells in the gastrointestinal (GI) tract. The results showed that activation of peripheral inflammasomes triggered NLRP3-mediated neuroinflammation in the brain [70][82]. In a study conducted, the gut microbiota from AD patients was transferred to APP/PSEN1 mice, causing microglial and NLRP3 inflammation leading to the release of inflammatory factors [71][83]. The GI tract then absorbs the inflammatory factors to cause inflammation. So, targeting the inflammasome signaling pathway through improving the composition of the gut microbiota would be a possible therapeutic option in AD.5
5.4. NETosis
Neutrophil extracellular traps (NETs), produced by innate immune phagocytes or neutrophils, participate in immune regulation and pathogen clearance [84]. The structure of NETs is a large, web-like structure built with cytosolic and granule proteins and decondensed chromatin [84]. Triggers such as ROS, antibodies and immune complexes, cytokines, pathogens (e.g., bacteria, fungi, protozoa, viruses), and bacterial cell wall structural moieties (e.g., LPS) can activate the formation of NETs [84,85]. There are two pathways through which NET formation can occur: NETosis and a non-lytic form of NETosis [85]. NETosis is the primary pathway through which NETs are formed. In this pathway, the neutrophil undergoes nuclear delobulation, including disintegration of the nuclear envelope, resulting in chromatin decondensation and rupture of the plasma membrane, and most importantly, the release of NETs [85]. The non-lytic form of NETosis avoids cell death; instead, the nuclear chromatin and granule proteins are released through secreted expulsion and degranulation, respectively. Then, in the extracellular space, the components assemble into NETs [85]. One of the crucial functions of NETs is their ability to regulate inflammatory cytokines through other immune cells [85]. In atherosclerosis, microscopic cholesterol crystals trigger the release of NETs activating TLR2 and TLR4 to transcribe IL-6 and pro-IL-1β in macrophages. These activated inflammatory cytokines increase the levels of myeloid cells present in the vicinity of atherosclerotic lesions [85,86]. When studying mice lacking neutrophil proteases necessary for NETosis, the results show these mice experience lower inflammation and form smaller atherosclerotic lesions [86]. Hence, NETs have the potential to aggravate inflammation in various diseases, including AD. Neutrophil hyperactivation is a central part of AD pathogenesis, along with neutrophil activation causing inflammation. In a study aimed to understand the influence of neutrophils in a transgenic (TG) AD mouse model, the positron emission tomography (PET) imaging showed neutrophil accumulation, increased production of cationic antimicrobial protein of molecular weight 37 kDa (CAP37), which is a neutrophil-produced molecule, and increased activation of microglia in the brain [87]. One of the most widely used animal models of AD is the penta-TG mouse model of familial AD (5xFAD) that overexpresses human APP with Swedish (K670N/M671L), Florida (I716V), and London (V717I) mutations as well as human PSEN1 with M146L and L286V mutations. Another most widely used animal model of AD is the triple TG mouse model of AD (3xTg-AD) that recapitulates both Aβ and neurofibrillary tangles (NFT) pathologies with incorporation of three mutations such as APP Swedish, microtubule-associated protein tau (MAPT) P301L, and PSEN1 M146V1 associated with FAD. A study using 5xFAD and 3xTg-AD, the widely used two TG mice models of AD, further showed evidence of neutrophil accumulation in areas with Aβ deposits [88]. The presence of neutrophil accumulation began before the onset of AD pathogenesis (microgliosis, tau phosphorylation, and cognitive deterioration). The deletion of neutrophils using an anti-Ly6G antibody significantly decreased the AD progression occurring in the model mice [88]. Neutrophil hyperactivation also has implications in causing inflammation in gut diseases such as IBD (e.g., Crohn’s disease and ulcerative colitis), CRC, and intestinal ischemia-reperfusion injury (IRI) [88].5.5. NETosis and Gut Microbiota
The gut microbiota also plays a role in the case of neutrophil-led inflammation. In acute mesenteric IRI, the translocation of gut bacteria results in multiple organ failure [89]. Colonized GF mice with complex gut microbiota were analyzed to understand whether NETosis was inhibited. This study also experimented on the relationship between neutrophil-led TLR4/Toll-IL-1R domain-containing adaptor-inducing IFN-β (TRIF) signaling and NETosis. Inhibiting neutrophils decreases LPS-triggered NETosis in the colonized mice [89]. Even though NETs are important in preventing infections, overactivation of NET formation can dysregulate the intestine epithelium barrier [90]. In another study, NETs were observed to support the attachment of enteropathogenic Escherichia coli (E. coli) and Shigatoxigenic E. coli to the intestinal mucosa [90]. The exploration of the influence of gut flora on NET formation is important to create therapeutic options for AD.6. Autophagy in AD
In AD brains, the main pathological changes are the deposition of Aβ plaques and intracellular NFTs made from hyperphosphorylated tau proteins [72][73][91,92]. The accumulation of Aβ plaques begins from the dysregulation of Aβ, which is modulated by autophagy [73][92]. Figure 6 showcases autophagy, which is a self-degradative process that plays a critical role in maintaining cellular homeostasis [74][75][93,94]. Autophagy is required to degrade the misfolded proteins present in the brain to prevent neurodegenerative diseases. Further studies have shown that the expression of autophagy-related proteins is downregulated in AD, implying the importance of autophagy upregulation as a part of treatments for AD [76][95].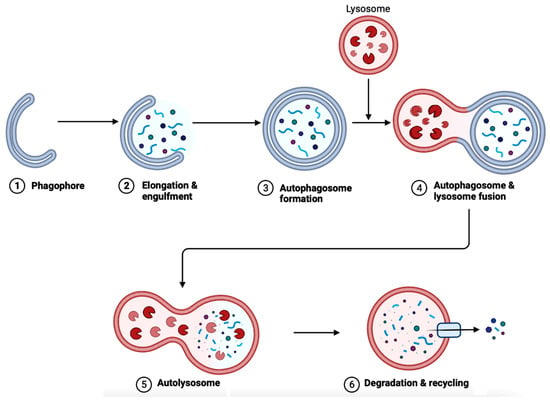
5.1. Implications of Gut Microbiota in Autophagy in AD
Implications of Gut Microbiota in Autophagy in AD
The gut microbiota has direct implications on many factors, including autophagy due to the existence of GBA. One of the ways to scrutinize whether gut flora has any effect on autophagy is to use GF mice. In the colonic epithelium of GF mice, the level of basal autophagy decreased compared to mice with intact gut flora [80][99]. The study also reinstated intestinal autophagy in vivo using butyrate-producing bacterial stain Butyrivibrio fibrisolvens. Bacteria-derived metabolites other than butyrate, such as indole-3-lactate produced by Lacticaseibacillus, Lactobacillus, Bifidobacterium, Megamonas, Roseburia, or Ruminococcus are also alternative options to induce intestinal autophagy [80][99]. These bacteria-derived metabolites can also modulate intestinal inflammation through the autophagy pathway. E. coli regulates autophagy through the NF-κB pathway, which upregulates selective microRNAs (miRNAs) before inhibiting ATG-specific proteins and then autophagy [81][100]. The NF-κB pathway is crucial to generating inflammatory responses; hence, maintaining the composition of the gut flora is important. Dysfunctional autophagy and gut dysbiosis are both in a positive feedback loop due to dysregulated autophagy resulting in impaired intestinal epithelial barrier function through altering the levels of expression of the CLDN2 (Claudin-2) gene that codes for the tight junction protein Claudin-2 in the intestinal mucosa [30][34]. Then, the increase in bacterial translocation causes gut dysbiosis [30][34]. Dysregulated autophagy causes gut dysbiosis and vice versa. This imbalance in the gut flora directly impacts autophagy in intestinal epithelial cells (IECs), such as paneth and goblet cells, epididymis epithelial cells (EECs), macrophages, dendritic cells (DCs), T and B cells, natural killer (NK) cells, and nerve cells of the enteric nervous system (ENS), and CNS. Gut dysbiosis also decreases the production of antimicrobial peptides (e.g., lysozyme, α-defensin, and phospholipase A2), intestinal epithelium regeneration, degrading bacterial accumulation, and pathogenic bacteria-led immune responses. Examining the neurons in Drosophila flies and mice, autophagy-deficient flies demonstrated a higher accumulation of ubiquitin/p62-positive protein inclusions and mitochondrial dysfunction, leading to cognitive deterioration [34]. A study analyzing in vitro and in vivo AD models along with brain tissue samples of AD patients showed sluggish autophagic flux with lower levels of degradation activity, resulting in the accumulation of autophagic vesicles [101]. Targeting gut microbiota would be realistic to decrease dysfunctional autophagy, which in turn would reduce the pathogenic features such as inflammation, oxidative stress, and accumulation of protein aggregates in many neurodegenerative diseases, including AD.67. The Bioflavonoids EGCG and GS as Therapeutic Agents for AD
6.1. Overview of Regulating Cell Signaling by EGCG and GS
7.1. Overview of Regulating Cell Signaling by EGCG and GS
These two bioflavonoids, EGCG and GS, have shown major influences on the NF-κB, mitogen-activated protein kinase (MAPK), epidermal growth factor receptor (EGFR), insulin-like growth factor (IGF), and mechanistic target of rapamycin (mTOR) signaling pathways to provide neuroprotection in neurodegenerative diseases (Table 23). Their mechanisms of action on these signaling pathways will be described below to show their therapeutic efficacies in AD.| Signaling Pathway in AD | Associated Functions | EGCG and GS | References |
|---|---|---|---|
| NF-κB pathway | Regulates pro-inflammatory genes | Both can inhibit the pathway | [29][73][74][75][76][33,92,93,94,95] |
| MAPK pathway | Regulates apoptosis, differentiation, etc. | Both can inhibit the pathway | [77][80][81][82][96[,9983,100,101],102] |
| EGFR pathway | Regulates gene expression and cell proliferation | Both can inhibit the pathway | [84][85][86][][91][103,104,10587][88][89,106],107[,10890,109,110] |
| IGF signal transduction pathway | Regulates cell differentiation, cell survival, and cell maintenance | Both can inhibit the pathway | [91][92]111[93][94][95][96][97],112[,11398,114][,11599][110,,116,117,118] |
| mTOR pathway | Regulates cell proliferation, apoptosis, and autophagy | Both can inhibit the pathway | [100][101][102][103][119[,120104,121,122],123] |
| 5-Hydroxytryptamine signaling pathway | Regulates serotonin production | Both can facilitate the pathway | [105][106][107][108][124,125,126,127] |