The immune system spontaneously recognizes and destroys foreign cells and organs when grafted into a genetically different individual. Organ transplantation is only successful because of the use of life-long immunosuppressive medications, which comes at the cost of severe toxicities. Thus, a major breakthrough in transplantation would be to be able to educate the immune system to accept grafted organs in the long term. A possible way to do that would be to exploit a physiological retro-control of the immune cells, which is based on the timely and coordinated expression of cell-surface receptors with inhibitory activities. In cancer, blocking these receptors (or Immune Checkpoints) boosts the anti-tumor functions of certain immune cells (the T-lymphocytes), with highly significant clinical benefits. Thus, it is likely that opposite actions, such as increasing the expression or the function of these receptors, would result in the dampening of the immune response against foreign organs.
- immune checkpoints
- organ transplantation
- tolerance
- immunotherapy
1. Introduction
2. The Role of Immune Checkpoints and Their Ligands in Solid Organ Transplantation
2.1. CTLA-4
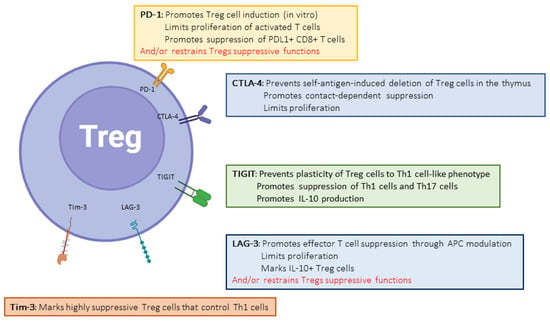
Cytotoxic T-Lymphocyte-Associated protein 4 (CTLA-4, or CD152) was the first described inhibitory molecule for T cells [19][8]. CTLA4 has gained incredible attention after the demonstration that blocking its interactions with its ligands can be highly beneficial to subsets of patients with cancer, paving the way for the immune therapy of cancer with immune checkpoint inhibitors (ICIs) [20,21][9][10]. The administration of such therapies in solid-organ-transplant patients was associated with a dramatic increase risk of acute, severe TCMR [22][11]. The paramount importance of CTLA-4 in regulating the immune system is demonstrated both by the phenotype of CTLA4 insufficiency (as well as LRBA deficiency, which inhibits CTLA4 expression by disrupting its recycling) in humans [13][12] and the early deaths of CTLA-4 knock-out (KO) mice [23][13]. CD80 (B7-1) and CD86 (B7-2) are CTLA-4-binding partners expressed on the surfaces of professional antigen-presenting cells. CTLA-4 functions at least at two separate levels: first, it is transiently expressed in activated T cells and disrupts CD28/B7 interactions, thereby acting as a physiological inhibitor of co-stimulation (Figure 1). In this respect, CTLA-4 is assumed to exert its regulatory effects at the time of T cell priming in lymphoid organs. Second, CTLA-4 is also strongly and constitutively expressed by Foxp3+-regulatory T cells (Tregs), and is crucial to their functions (Figure 2) [24][14]. This dual action of CTLA-4 is a source of complexity and difficulties to disentangle the cellular mechanisms at play in specific conditions [25][15]. This roadblock has been partially resolved through the generation of cell-type-specific and conditional KO mice. Indeed, the Treg-specific deletion of CTLA-4 is sufficient for mice to develop a fatal lymphoproliferative disease, showing the crucial role of CTLA-4 for the Treg-mediated regulation of conventional T (Tconv) cells in vivo [26][16]. On the other hand, two groups developed conditional KO to study the consequences of CTLA deletion at an adult age [27,28][17][18].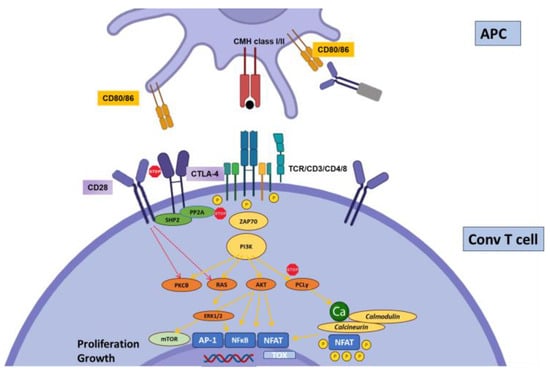
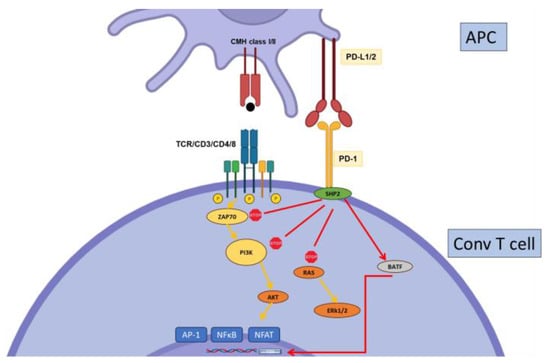
2.2. PD-1
2.3. Tim-3
Constitutively expressed by dendritic cells and regulatory T cells [57][29], T-cell-immunoglobulin-and-mucin-containing protein-3 (Tim-3, or CD366) is induced early after activation on conventional T cells [58][30]. Tim-3 expression by Tregs may play a major role in their suppressive function, at least in specific contexts [59,60][31][32]. In contrast to CTLA-4 or PD-1, Tim-3 expression is confined to the Th1 and Th17 cell subsets of CD4+ T cells, and to IFNγ-producing -CD8+ T cells [58,61,62][30][33][34]. These observations suggest a specific role for Tim-3 in the regulation of the more inflammatory subsets of T cells. Another peculiarity of Tim-3 is the existence of several ligands, including galectin-9 (Gal9), Carcinoembryonic-antigen-related cell-adhesion molecule 1(CEACAM-1), Phosphatidylserine (PtdSer), and high-mobility group box 1 (HMGB1), showing wide cellular and tissue expression. However, Gal9 seems to be the major Tim-3 ligand. Tim-3 signaling directly interferes with the CD3 signaling cascade. However, the mechanisms by which Tim-3 regulates T cell functions are poorly understood; the current hypotheses propose that Tim-3 displays a dual stimulatory/inhibitory function depending on the interaction with an intra-cytoplasmic signaling adaptor, Bat3 (Figure 4).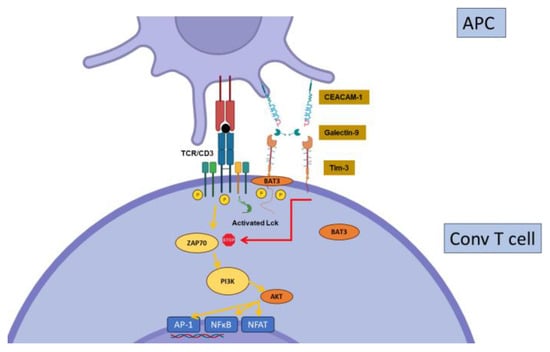
2.4. BTLA
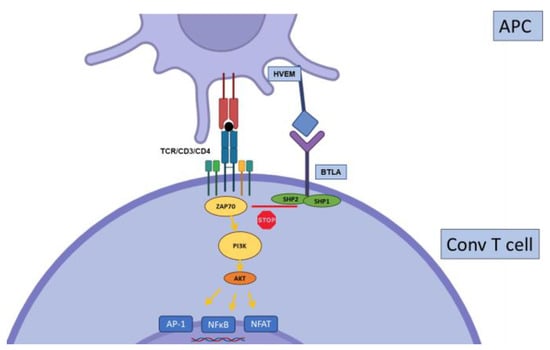
2.5. TIGIT
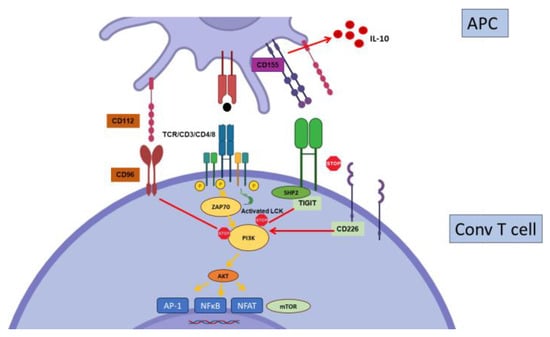
2.6. CD244
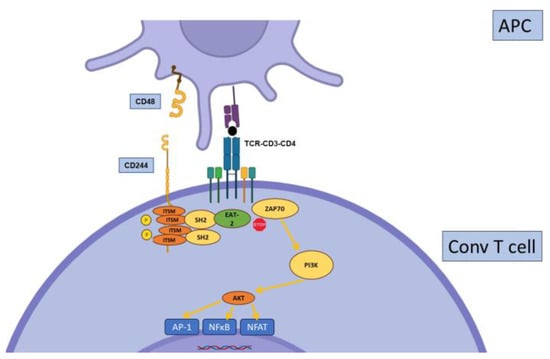
2.7. Other Inhibitory Receptors
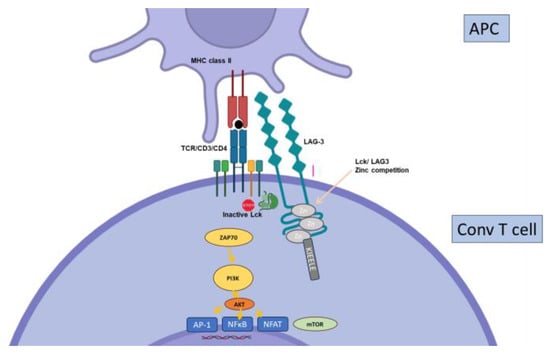
3. T Cell Exhaustion in Solid Organ Transplantation
4. Interactions between Immunosuppressive Drugs and IC Expression and Function
It is highly likely that immunosuppressive drugs may have an impact on IC expression and T cell exhaustion. In the mouse model of chronic LCMV infection (a model for antigen-specific exhaustion), a complex interplay between PD-1 and mTOR is demonstrated, whereby rapamycin inhibits the effect of PD-1 blockade [120][61]. This suggests that mTOR regulates T cell exhaustion in this model. However, blocking mTOR at the beginning of a T cell response increases the response and inhibits exhaustion [121][62]. The chronic administration of rapamycin to healthy aged mice decreases the expression of PD-1 and LAG-3 and increases proliferative capacities, suggesting the reduction of “natural” exhaustion [122][63]. On the other hand, another widely used IS agent, mycophenolate mofetil (MMF), may show an opposite effect. Several co-IRs (PD-1, LAG3, TIGIT…) displayed higher expression on blood Treg cells from stable liver transplant recipients treated with tacrolimus (a CNi) + MMF compared to patients treated with tacrolimus only, suggesting that MMF increases IR expression [128][64]. Further, Tregs from patients treated with the same regimen were more effective at inhibiting T cell proliferation [129][65], and this effect was partially abrogated when treated with a combination of anti-TIGIT and anti-PD-1.5. Harnessing Inhibitory Pathways in Solid Organ Transplantation
Beyond the potential role of ICs in predicting the outcomes, several molecules harnessing ICs are currently in the pipe. The success of IC blockade in subsets of cancer patients has prompted a major endeavor in both academic and industry groups to develop new strategies to overcome T cell exhaustion. The reverse action may be efficient in organ transplantation. Indeed, targeting co-stimulatory pathways such as CD80/86 with belatacept has demonstrated interesting results with respect to the prevention of acute and chronic allograft rejections. It is noteworthy that most murine studies showing the role of ICs such as PD-1 in regulating chronic organ rejection have been performed alongside the blockading of co-stimulatory pathways; the manipulation of inhibitory pathways in the presence of intact co-stimulatory signaling has often yielded disappointing results. This observation may simply reflect the fact that blocking co-stimulation while enhancing inhibition is probably more efficient for the regulation of T cell activation as compared with targeting only one of these pathways. However, it is important to recall that insufficient help from CD4+ is one of the processes leading to CD8+ T cell exhaustion [106,130][66][67]. Thus, combining IC activation with co-stimulation blockade may be a synergistic approach because the latter would induce or contribute to differentiating T cells towards a hypo-responsive, exhausted state.References
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004.
- Le Mercier, I.; Lines, J.L.; Noelle, R.J. Beyond CTLA-4 and PD-1, the Generation Z of Negative Checkpoint Regulators. Front. Immunol. 2015, 6, 418.
- Ziogas, D.C.; Theocharopoulos, C.; Lialios, P.P.; Foteinou, D.; Koumprentziotis, I.A.; Xynos, G.; Gogas, H. Beyond CTLA-4 and PD-1 Inhibition: Novel Immune Checkpoint Molecules for Melanoma Treatment. Cancers 2023, 15, 2718.
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495.
- Franco, F.; Jaccard, A.; Romero, P.; Yu, Y.R.; Ho, P.C. Metabolic and epigenetic regulation of T-cell exhaustion. Nat. Metab. 2020, 2, 1001–1012.
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801.
- Boumaza, X.; Bonneau, B.; Roos-Weil, D.; Pinnetti, C.; Rauer, S.; Nitsch, L.; Del Bello, A.; Jelcic, I.; Suhs, K.W.; Gasnault, J.; et al. Progressive Multifocal Leukoencephalopathy Treated by Immune Checkpoint Inhibitors. Ann. Neurol. 2023, 93, 257–270.
- Brunet, J.F.; Denizot, F.; Luciani, M.F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.G.; Golstein, P. A new member of the immunoglobulin superfamily—CTLA-4. Nature 1987, 328, 267–270.
- Egen, J.G.; Kuhns, M.S.; Allison, J.P. CTLA-4: New insights into its biological function and use in tumor immunotherapy. Nat. Immunol. 2002, 3, 611–618.
- Phan, G.Q.; Yang, J.C.; Sherry, R.M.; Hwu, P.; Topalian, S.L.; Schwartzentruber, D.J.; Restifo, N.P.; Haworth, L.R.; Seipp, C.A.; Freezer, L.J.; et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 2003, 100, 8372–8377.
- Nguyen, L.S.; Ortuno, S.; Lebrun-Vignes, B.; Johnson, D.B.; Moslehi, J.J.; Hertig, A.; Salem, J.E. Transplant rejections associated with immune checkpoint inhibitors: A pharmacovigilance study and systematic literature review. Eur. J. Cancer 2021, 148, 36–47.
- Schubert, D.; Bode, C.; Kenefeck, R.; Hou, T.Z.; Wing, J.B.; Kennedy, A.; Bulashevska, A.; Petersen, B.S.; Schaffer, A.A.; Gruning, B.A.; et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat. Med. 2014, 20, 1410–1416.
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547.
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-mediated inhibition in regulation of T cell responses: Mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594.
- Jain, N.; Nguyen, H.; Chambers, C.; Kang, J. Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc. Natl. Acad. Sci. USA 2010, 107, 1524–1528.
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275.
- Paterson, A.M.; Lovitch, S.B.; Sage, P.T.; Juneja, V.R.; Lee, Y.; Trombley, J.D.; Arancibia-Carcamo, C.V.; Sobel, R.A.; Rudensky, A.Y.; Kuchroo, V.K.; et al. Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J. Exp. Med. 2015, 212, 1603–1621.
- Klocke, K.; Sakaguchi, S.; Holmdahl, R.; Wing, K. Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood. Proc. Natl. Acad. Sci. USA 2016, 113, E2383–E2392.
- Poirier, N.; Azimzadeh, A.M.; Zhang, T.; Dilek, N.; Mary, C.; Nguyen, B.; Tillou, X.; Wu, G.; Reneaudin, K.; Hervouet, J.; et al. Inducing CTLA-4-dependent immune regulation by selective CD28 blockade promotes regulatory T cells in organ transplantation. Sci. Transl. Med. 2010, 2, 17ra10.
- Perez-Garcia, A.; De la Camara, R.; Roman-Gomez, J.; Jimenez-Velasco, A.; Encuentra, M.; Nieto, J.B.; de la Rubia, J.; Urbano-Ispizua, A.; Brunet, S.; Iriondo, A.; et al. CTLA-4 polymorphisms and clinical outcome after allogeneic stem cell transplantation from HLA-identical sibling donors. Blood 2007, 110, 461–467.
- Santiago, J.L.; Sanchez-Perez, L.; Perez-Flores, I.; de la Higuera, M.A.M.; Romero, N.C.; Querol-Garcia, J.; Urcelay, E.; Sanchez-Fructuoso, A.I. Association of Polymorphisms in T-Cell Activation Costimulatory/Inhibitory Signal Genes with Allograft Kidney Rejection Risk. Front. Immunol. 2021, 12, 650979.
- Rosik, J.; Szostak, B.; Machaj, F.; Pawlik, A. The Role of CTLA4 and Its Polymorphisms in Solid Organ and Haematopoietic Stem Cell Transplantation. Int. J. Mol. Sci. 2021, 22, 3081.
- Ozkaynak, E.; Wang, L.; Goodearl, A.; McDonald, K.; Qin, S.; O′Keefe, T.; Duong, T.; Smith, T.; Gutierrez-Ramos, J.C.; Rottman, J.B.; et al. Programmed death-1 targeting can promote allograft survival. J. Immunol. 2002, 169, 6546–6553.
- Ito, T.; Ueno, T.; Clarkson, M.R.; Yuan, X.; Jurewicz, M.M.; Yagita, H.; Azuma, M.; Sharpe, A.H.; Auchincloss, H., Jr.; Sayegh, M.H.; et al. Analysis of the role of negative T cell costimulatory pathways in CD4 and CD8 T cell-mediated alloimmune responses in vivo. J. Immunol. 2005, 174, 6648–6656.
- Riella, L.V.; Watanabe, T.; Sage, P.T.; Yang, J.; Yeung, M.; Azzi, J.; Vanguri, V.; Chandraker, A.; Sharpe, A.H.; Sayegh, M.H.; et al. Essential role of PDL1 expression on nonhematopoietic donor cells in acquired tolerance to vascularized cardiac allografts. Am. J. Transplant. 2011, 11, 832–840.
- Koga, N.; Suzuki, J.; Kosuge, H.; Haraguchi, G.; Onai, Y.; Futamatsu, H.; Maejima, Y.; Gotoh, R.; Saiki, H.; Tsushima, F.; et al. Blockade of the interaction between PD-1 and PD-L1 accelerates graft arterial disease in cardiac allografts. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2057–2062.
- Luo, Z.; Liao, T.; Zhang, Y.; Zheng, H.; Sun, Q.; Han, F.; Ma, M.; Ye, Y.; Sun, Q. Ex vivo anchored PD-L1 functionally prevent in vivo renal allograft rejection. Bioeng. Transl. Med. 2022, 7, e10316.
- Starke, A.; Lindenmeyer, M.T.; Segerer, S.; Neusser, M.A.; Rusi, B.; Schmid, D.M.; Cohen, C.D.; Wuthrich, R.P.; Fehr, T.; Waeckerle-Men, Y. Renal tubular PD-L1 (CD274) suppresses alloreactive human T-cell responses. Kidney Int. 2010, 78, 38–47.
- Boenisch, O.; D’Addio, F.; Watanabe, T.; Elyaman, W.; Magee, C.N.; Yeung, M.Y.; Padera, R.F.; Rodig, S.J.; Murayama, T.; Tanaka, K.; et al. TIM-3: A novel regulatory molecule of alloimmune activation. J. Immunol. 2010, 185, 5806–5819.
- Hastings, W.D.; Anderson, D.E.; Kassam, N.; Koguchi, K.; Greenfield, E.A.; Kent, S.C.; Zheng, X.X.; Strom, T.B.; Hafler, D.A.; Kuchroo, V.K. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur. J. Immunol. 2009, 39, 2492–2501.
- Gupta, S.; Thornley, T.B.; Gao, W.; Larocca, R.; Turka, L.A.; Kuchroo, V.K.; Strom, T.B. Allograft rejection is restrained by short-lived TIM-3+PD-1+Foxp3+ Tregs. J. Clin. Investig. 2012, 122, 2395–2404.
- Yan, J.; Zhang, Y.; Zhang, J.P.; Liang, J.; Li, L.; Zheng, L. Tim-3 expression defines regulatory T cells in human tumors. PLoS ONE 2013, 8, e58006.
- Tang, R.; Rangachari, M.; Kuchroo, V.K. Tim-3: A co-receptor with diverse roles in T cell exhaustion and tolerance. Semin. Immunol. 2019, 42, 101302.
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541.
- Sanchez-Fueyo, A.; Tian, J.; Picarella, D.; Domenig, C.; Zheng, X.X.; Sabatos, C.A.; Manlongat, N.; Bender, O.; Kamradt, T.; Kuchroo, V.K.; et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 2003, 4, 1093–1101.
- Ning, Z.; Liu, K.; Xiong, H. Roles of BTLA in Immunity and Immune Disorders. Front. Immunol. 2021, 12, 654960.
- Tao, R.; Wang, L.; Han, R.; Wang, T.; Ye, Q.; Honjo, T.; Murphy, T.L.; Murphy, K.M.; Hancock, W.W. Differential effects of B and T lymphocyte attenuator and programmed death-1 on acceptance of partially versus fully MHC-mismatched cardiac allografts. J. Immunol. 2005, 175, 5774–5782.
- Zhang, J.; Zhang, H.; Wang, Z.; Yang, H.; Chen, H.; Cheng, H.; Zhou, J.; Zheng, M.; Tan, R.; Gu, M. BTLA suppress acute rejection via regulating TCR downstream signals and cytokines production in kidney transplantation and prolonged allografts survival. Sci. Rep. 2019, 9, 12154.
- Zhang, H.; Wang, Z.; Zhang, J.; Gui, Z.; Han, Z.; Tao, J.; Chen, H.; Sun, L.; Fei, S.; Yang, H.; et al. Combined Immunotherapy With Belatacept and BTLA Overexpression Attenuates Acute Rejection Following Kidney Transplantation. Front. Immunol. 2021, 12, 618737.
- Joller, N.; Hafler, J.P.; Brynedal, B.; Kassam, N.; Spoerl, S.; Levin, S.D.; Sharpe, A.H.; Kuchroo, V.K. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J. Immunol. 2011, 186, 1338–1342.
- Zhao, J.; Li, L.; Yin, H.; Feng, X.; Lu, Q. TIGIT: An emerging immune checkpoint target for immunotherapy in autoimmune disease and cancer. Int. Immunopharmacol. 2023, 120, 110358.
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57.
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863.
- Lozano, E.; Dominguez-Villar, M.; Kuchroo, V.; Hafler, D.A. The TIGIT/CD226 axis regulates human T cell function. J. Immunol. 2012, 188, 3869–3875.
- McNerney, M.E.; Lee, K.M.; Kumar, V. 2B4 (CD244) is a non-MHC binding receptor with multiple functions on natural killer cells and CD8+ T cells. Mol. Immunol. 2005, 42, 489–494.
- Vaidya, S.V.; Mathew, P.A. Of mice and men: Different functions of the murine and human 2B4 (CD244) receptor on NK cells. Immunol. Lett. 2006, 105, 180–184.
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405.
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513.
- Zhang, Q.; Chikina, M.; Szymczak-Workman, A.L.; Horne, W.; Kolls, J.K.; Vignali, K.M.; Normolle, D.; Bettini, M.; Workman, C.J.; Vignali, D.A.A. LAG3 limits regulatory T cell proliferation and function in autoimmune diabetes. Sci. Immunol. 2017, 2, eaah4569.
- Aggarwal, V.; Workman, C.J.; Vignali, D.A.A. LAG-3 as the third checkpoint inhibitor. Nat. Immunol. 2023, 24, 1415–1422.
- Sega, E.I.; Leveson-Gower, D.B.; Florek, M.; Schneidawind, D.; Luong, R.H.; Negrin, R.S. Role of lymphocyte activation gene-3 (Lag-3) in conventional and regulatory T cell function in allogeneic transplantation. PLoS ONE 2014, 9, e86551.
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687.
- Collier, J.L.; Weiss, S.A.; Pauken, K.E.; Sen, D.R.; Sharpe, A.H. Not-so-opposite ends of the spectrum: CD8(+) T cell dysfunction across chronic infection, cancer and autoimmunity. Nat. Immunol. 2021, 22, 809–819.
- Ghobrial, I.I.; Morris, A.G.; Booth, L.J. Clinical significance of in vitro donor-specific hyporesponsiveness in renal allograft recipients as demonstrated by the MLR. Transpl. Int. 1994, 7, 420–427.
- Mason, P.D.; Robinson, C.M.; Lechler, R.I. Detection of donor-specific hyporesponsiveness following late failure of human renal allografts. Kidney Int. 1996, 50, 1019–1025.
- Bestard, O.; Nickel, P.; Cruzado, J.M.; Schoenemann, C.; Boenisch, O.; Sefrin, A.; Grinyo, J.M.; Volk, H.D.; Reinke, P. Circulating alloreactive T cells correlate with graft function in longstanding renal transplant recipients. J. Am. Soc. Nephrol. 2008, 19, 1419–1429.
- Del Bello, A.; Gouin, A.; Chaubet, C.; Kamar, N.; Treiner, E. The CD226/TIGIT axis is involved in T cell hypo-responsiveness appearance in long-term kidney transplant recipients. Sci. Rep. 2022, 12, 11821.
- Game, D.S.; Hernandez-Fuentes, M.P.; Chaudhry, A.N.; Lechler, R.I. CD4+CD25+ regulatory T cells do not significantly contribute to direct pathway hyporesponsiveness in stable renal transplant patients. J. Am. Soc. Nephrol. 2003, 14, 1652–1661.
- van der List, A.C.J.; Litjens, N.H.R.; Klepper, M.; Prevoo, F.; Betjes, M.G.H. Progressive Loss of Donor-Reactive CD4(+) Effector Memory T Cells due to Apoptosis Underlies Donor-Specific Hyporesponsiveness in Stable Renal Transplant Recipients. J. Immunol. 2022, 209, 1389–1400.
- Sarraj, B.; Ye, J.; Akl, A.I.; Chen, G.; Wang, J.J.; Zhang, Z.; Abadja, F.; Abecassis, M.; Miller, S.D.; Kansas, G.S.; et al. Impaired selectin-dependent leukocyte recruitment induces T-cell exhaustion and prevents chronic allograft vasculopathy and rejection. Proc. Natl. Acad. Sci. USA 2014, 111, 12145–12150.
- Staron, M.M.; Gray, S.M.; Marshall, H.D.; Parish, I.A.; Chen, J.H.; Perry, C.J.; Cui, G.; Li, M.O.; Kaech, S.M. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity 2014, 41, 802–814.
- Ando, S.; Perkins, C.M.; Sajiki, Y.; Chastain, C.; Valanparambil, R.M.; Wieland, A.; Hudson, W.H.; Hashimoto, M.; Ramalingam, S.S.; Freeman, G.J.; et al. mTOR regulates T cell exhaustion and PD-1-targeted immunotherapy response during chronic viral infection. J. Clin. Investig. 2023, 133, e160025.
- Hurez, V.; Dao, V.; Liu, A.; Pandeswara, S.; Gelfond, J.; Sun, L.; Bergman, M.; Orihuela, C.J.; Galvan, V.; Padron, A.; et al. Chronic mTOR inhibition in mice with rapamycin alters T, B, myeloid, and innate lymphoid cells and gut flora and prolongs life of immune-deficient mice. Aging Cell 2015, 14, 945–956.
- Zeng, Q.; Yuan, X.Y.; Li, W.; Liu, B.W.; Zhao, X.; Ren, G.J.; Wang, Y.; Dou, J.; Wang, G.Y. Effects of tacrolimus (FK506) and mycophenolate mofetil (MMF) on regulatory T cells and co-inhibitory receptors in the peripheral blood of human liver allograft patients. Immunopharmacol. Immunotoxicol. 2019, 41, 380–385.
- Zeng, Q.; Yuan, X.; Cao, J.; Zhao, X.; Wang, Y.; Liu, B.; Liu, W.; Zhu, Z.; Dou, J. Mycophenolate mofetil enhances the effects of tacrolimus on the inhibitory function of regulatory T cells in patients after liver transplantation via PD-1 and TIGIT receptors. Immunopharmacol. Immunotoxicol. 2021, 43, 239–246.
- Lu, Y.J.; Barreira-Silva, P.; Boyce, S.; Powers, J.; Cavallo, K.; Behar, S.M. CD4 T cell help prevents CD8 T cell exhaustion and promotes control of Mycobacterium tuberculosis infection. Cell Rep. 2021, 36, 109696.
- Aubert, R.D.; Kamphorst, A.O.; Sarkar, S.; Vezys, V.; Ha, S.J.; Barber, D.L.; Ye, L.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. Proc. Natl. Acad. Sci. USA 2011, 108, 21182–21187.