Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Claudiu Morgovan and Version 2 by Camila Xu.
Heart failure (HF) with preserved ejection fraction (HFpEF) is an increasingly frequent form and is estimated to be the dominant form of HF. On the other hand, HFpEF is a syndrome with systemic involvement, and it is characterized by multiple cardiac and extracardiac pathophysiological alterations.
- heart failure
- HFpEF
- pathophysiological mechanism
- phenotypes
- treatment
1. Introduction
The diagnosis and treatment of heart failure (HF) with preserved ejection fraction (HFpEF) represent one of the greatest challenges for physicians today.
Although HFpEF has been seen as a mild condition in terms of organ damage, when it comes to treating these patients, there has been limited progress in developing an effective therapy. Perhaps this situation is due to the fact that HFpEF is a systemic syndrome with multi-organ involvement, which corroborates multiple cardiac and extracardiac physiopathological alterations [1][2][3][3,4,5].
HFpEF was defined by Dr. Luchi et al. in 1982, being the first group of researchers to describe typical heart failure symptoms in a group of patients with preserved left ventricular (LV) ejection fraction (EF) [4][6]. Recently, the European Society of Cardiology (ESC) united under the term HFpEF patients with preserved left ventricular EF (LVEF ≥ 50%) but with evidence of diastolic dysfunction or structural heart disease, classic signs and symptoms of heart failure and elevated plasma natriuretic peptide (NP) levels [5][1].
As with any definition of a syndrome, there are limitations, one of which is related to the lack of congestion in compensated HF. Another limitation is represented by the group of patients with HF symptoms who present abnormal hemodynamics exclusively during physical exercises [6][7]. HFpEF has been seen as a low-impact condition, yet patients have symptoms, signs, and quality of life not much different from those of patients with heart failure with reduced ejection fraction (HFrEF). Expert opinions support that HFpEF is rather a heterogeneous syndrome that includes different phenotypes with a spectrum of distinct, overlapping characteristics [7][8]. The etiology of HFpEF is unclear, and probably often multifactorial, but several culprits have been identified: microvascular lesions, low-grade systemic inflammation, and general oxidative stress (evolving in the context of comorbidities associated with endothelial dysfunction), all of these leading to myocardial remodeling and fibrosis [8][9]. These detrimental elements seem to participate fundamentally in the pathogenesis of the disease [7][8].
2. Pathophysiology of HFpEF
2.1. Left Ventricular Structure and Remodeling
The initial model indicated for HFpEF in descriptive studies was that of a ventricle of normal size but with hypertrophied walls (concentric left ventricular hypertrophy) [9][55]. Like any model, this one was not representative of all patients with HFpEF, some of them not having cardiac structural remodeling, the left ventricular geometry being normal. However, most patients with HFpEF correspond to the previously exposed pattern, recognized by the following characteristics: hypertrophy by increasing left ventricle (LV) wall thickness and/or LV mass and end-diastolic volume within normal or near-normal limits. Hypertrophy can be concentric hypertrophy by increasing the ratio of myocardial mass to cavity volume or generalized hypertrophy by increasing relative wall thickness (RWT) [10][56]. There is also a group of patients who present eccentric hypertrophy, and their proportion can reach 16% [11][57]. Going further, at the microscopic level, there are differences in the structure of the cardiomyocytes of patients with HFpEF compared to patients with HFrEF, with the cardiomyocytes of patients with HFpEF being thicker and less elongated [9][55]. However, according to Dao-Fu Dai et al., the fact that there is an age-dependent increase in the thickness of the left ventricular wall must be taken into account. This was shown by the analyses from the Framingham Heart Study and the Baltimore Longitudinal Study on Aging, studies that investigated apparently healthy adults using cardiac ultrasound. The conclusion was that there was an increased prevalence of left ventricular hypertrophy with age in both men and women, even in the absence of clinical hypertension, the most common risk factor for CVD [12][58]. And as aging is one of the most important contributors to HFpEF, left ventricular hypertrophy can be present but not necessarily in relation to HFpEF [12][58].2.2. Left Ventricular Diastolic Dysfunction
The generally accepted definition of diastolic dysfunction states that the left ventricle is unable to fill to an adequate level, correlated to the body’s needs (end-diastolic volume—EDV) under conditions of low (but normal) pressure [13][59]. Although initially HFpEF was named as HF with diastolic dysfunction, diastolic dysfunction is not superimposed with HFpEF [13][59]. As Borlaug BA et al. state, diastolic dysfunction is independent of normal ejection fraction of the left ventricle. Diastolic dysfunction is the result of an abnormal distensibility of the LV that results in reduced relaxation and filling, regardless of whether or not the contractile function of the LV is normal, or whether or not these abnormalities produce symptoms [13][59]. It is accepted that diastolic dysfunction is part of normal human aging. The fact is reinforced by its detection in many people who do not have or will never have HFpEF. This occurs as a result of reduced filling of the LV in early diastole. This phenomenon becomes visible with increasing age in both sexes, through the decrease in ventricular elasticity due to the fibrosis of the LV walls and through the delay in active ventricular relaxation. Several mechanisms contribute to diastolic dysfunction like delayed relaxation due to reducing the efficiency of calcium capture and retention in the myocardial sarcoplasmic reticulum (SERCA2a), thus contributing to low sucking capability of the ventricle and wall rigidity. The development of diastolic dysfunction, in patients with HFpEF, does not affect the final filling volume of the LV, but this filling is difficult, as abnormally high filling pressures are required [9][55].2.3. Ventricular Dyssynchrony
Ventricular dyssynchrony is defined as an increase in the time difference between the contractions of the two ventricles. The desynchronization of the moment of contraction of the two ventricles reduces the cardiac efficiency in performing the contraction and relaxation of the myocardium and can be correlated with the occurrence of HF [14][60]. It is known that cardiac dyssynchrony is, in the case of HFrEF, associated with a higher risk of adverse outcomes [15][61]. Although the electrical asynchrony in the case of HFpEF does not have as its main mechanism the bundle-branch block as in the case of HFrEF, these patients nevertheless tend to have wider QRS complexes, in these conditions, mechanical systolic and diastolic asynchrony being quite frequent [14][16][60,62]. To assess dyssynchrony, 2D speckle-tracking echocardiography (STE) is used to calculate global longitudinal strain. The advantage over the Doppler evaluation is its angle independence [17][63]. Patients with HFpEF have greater ventricular dyssynchrony compared to healthy people; this was expected, but dyssynchrony exists even in patients with a narrow QRS complex and LVEF ≥ 55% [16][62]. In HFpEF, mechanical dyssynchrony depends on QRS width (electrical dyssynchrony), ventricular hypertrophy, and diastolic but not systolic dysfunction [16][62].2.4. Atrial Dysfunction and Atrial Fibrillation
Atrial fibrillation is a more frequent pathology in patients with HFpEF compared to those with HFrEF [18][64]. The left atrium (LA) functions as a buffer, absolutely necessary between the pulmonary veins and the LV. It is characterized by four functions. Two of them are passive and aim at blood storage, the reservoir function, respectively blood transfer into the LV, the conduit function. The other two are active, being represented by the accumulation of potential energy in the form of pressure, the battery function, respectively the contractile function that increases LV stroke output [19][20][65,66]. Studies have shown that once HFpEF is developed even at an early stage, patients need left atrium contraction to achieve normal LV filling compared to healthy individuals [19][65]. To maintain adequate LV filling, the atrial contribution, through contraction, becomes increasingly important with aging, but sustained atrial contraction also increases atrial pressure, which leads over time to atrial hypertrophy, which is a risk factor for atrial fibrillation [12][58]. The more important factor is the contractile function of the left atrium as patients with HFpEF, who develop atrial fibrillation, have a reduced quality of life due to reduced exercise capacity and the development or worsening of right ventricular (RV) dysfunction and increased mortality regardless of the severity of HF [21][67]. Melenovsky et al. found that the occurrence of atrial fibrillation in patients with HFpEF, compared to those in sinus rhythm, was associated with greater hemodynamic suffering of the right heart manifested by higher pressures in the pulmonary artery (PA), dilation and functional alteration of the right cavities [22][68].2.5. Right Ventricle Dysfunction (RVD) and Pulmonary Vascular Disease
HfpEF is frequently associated with pulmonary hypertension (PH), right ventricular disease being part of this constellation of diseases included in the HfpEF syndrome. Up to two-thirds of patients with HfpEF present simultaneously PH [23][69]. Beyond the simple presence of PH, its severity is very important for a patient’s prognosis, as it is known that there is an increase in the relative risk of mortality of 28% for every 10 mmHg increase in PA pressure [24][70]. Reducing pulmonary arterial pressure, by using diuretics, decreases the number of hospitalizations for decompensated HFpEF [24][70]. RVD in most cases is caused by impaired pulmonary circulation, the most frequent cause being PH. Any increase in pulmonary arterial resistance requires an increase in myocardial contractility from the RV side, this increase being up to five times compared to the normal hemodynamic situation [25][71]. The problem is not that the afterload increases, but that it increases persistently, the RV not being able to cope with an increased pressure regime for long periods, given that the thickness of the RV wall is much reduced compared to that of the LV. As a result, the persistence of an increased arterial resistance will lead to RV dilation, decreased myocardial contractility (ventriculo-arterial decoupling), and decreased RV ejection fraction (RVEF). As a consequence, the RV cannot maintain an adequate cardiac output, resulting in the clinical appearance of HF. Statistical data show that RVD can be found in up to 50% of patients with HFpEF, the development of RVD in patients with PH being a strong marker of increased morbidity and mortality, independent of the severity of HF [26][72].2.6. Pericardial Restraint
The pericardial sac contributes to the good functioning of the heart through multiple roles, one of them being the limitation of the distension of the ventricular filling; this correlated with the venous return contributing to the increase in the intracardiac pressure [27][73]. Increased filling pressure in the LV is responsible for most of the symptoms in HFpEF. These elevated LV filling pressures occur in HFpEF predominantly due to myocardial relaxation abnormalities, but an important contribution also comes from pericardial constriction [28][74]. In an attempt to reduce intraventricular pressure, experiments were performed on animals in which it was demonstrated that pericardial resection reduces the increase in LV filling pressures in conditions of volume overload, both in normal hearts and those with diastolic dysfunction [27][73]. There are two phenotypes of HFpEF in which the pericardium contributes to the development and worsening of HF, the pulmonary hypertension phenotype and the obesity (cardiometabolic) phenotype [29][30][75,76]. In the case of patients with PH, it is understandable that the tension of the pericardial sac leads to an increase in RV pressure in exaggerated afterload conditions. In the obese phenotype, the combination of excess pericardial fat and increased cardiac volume leads to exaggeration of pericardial restraint [28][74].2.7. Vascular Stiffness and Endothelial Dysfunction
Patients diagnosed with HFpEF frequently associate with reduced central aortic compliance and increased peripheral arterial stiffness [31][77]. This was expected as long as the increase in arterial stiffness is associated with diastolic dysfunction, with more arterial stiffness being responsible for the accelerated development of diastolic dysfunction [32][78]. Beyond the increased arterial stiffness, a sign of systemic vascular dysfunction, more than 70% of patients with HFpEF also associate with a functional coronary impairment manifested by a reduced coronary myocardial flow reserve [33][79]. The common element of the two vascular alterations (micro- and macro-vascular) is endothelial dysfunction. The same endothelial dysfunction was found in patients with HFpEF [34][80]. It is demonstrated that the presence and severity of endothelial dysfunction in patients with HFpEF contribute to a worse prognosis due to higher rates of acute cardiovascular events, a worse quality of life due to more severe symptoms, and reduced exercise capacity [31][77] (Figure 12).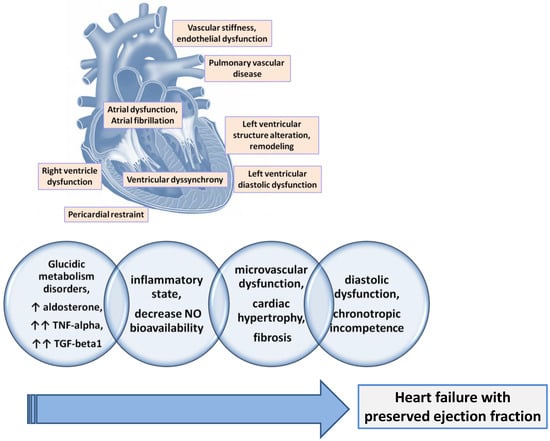
Figure 12.
The main physiopathological mechanisms involved in the development of HFpEF.