Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Lindsay Dong and Version 1 by Tadayuki Komori.
Studies point to the involvement of endolysosomal defects in parkinson’s disease (PD). The endolysosomal system, which tightly controls a flow of endocytosed vesicles targeted either for degradation or recycling, is regulated by a number of Rab GTPases. Their associations with leucine-rich repeat kinase 2 (LRRK2), a major causative and risk protein of PD, has also been one of the hot topics in the field.
- LRRK2
- Rab
- endolysosome
- intracellular transport
- Parkinson’s disease
1. Introduction
Since its discovery as the protein responsible for Parkinson’s disease (PD) in the PARK8 locus in 2004 [1[1][2],2], leucine-rich repeat kinase 2 (LRRK2) has been one of the main focus molecules associated with this neurodegenerative disease. The physiological roles of LRRK2 have been linked to a myriad of cellular processes, such as several types of autophagy including macroautophagy and mitophagy [3[3][4],4], endocytosis and intracellular transport involving the trans-Golgi network (TGN) and other organelles [5[5][6][7][8],6,7,8], the regulation of microtubules [5[5][6],6], interaction with bacterial pathogens [7], regulation of lysosomal homeostasis [8], and much more.
The pathogenic features of LRRK2 have also been studied by analyzing mutations associated with PD, and most, if not all, mutations point towards a similar effect: augmentation of substrate phosphorylation [8,9][8][9]. Albeit these findings, the exact mechanism of how defects in LRRK2 lead to PD has been elusive for nearly a score of years now. An auspicious approach to this enigma would be its link to the endolysosomal system and their regulators, Rab GTPases, as a considerable number of findings indicate connections between these [10].
Rab GTPases bind to membranes and utilize their affinity change via their guanine nucleotide binding status to form specific functional domains on their corresponding organelle membranes [11], hence called the master regulators of intracellular vesicular traffic. Some of the first reports that related Rab GTPases to PD were around 20 years ago, when α-synuclein was reported to interact with several Rab GTPases [12] or α-synuclein-induced neuronal loss was rescued by overexpression of Rab1 [13].
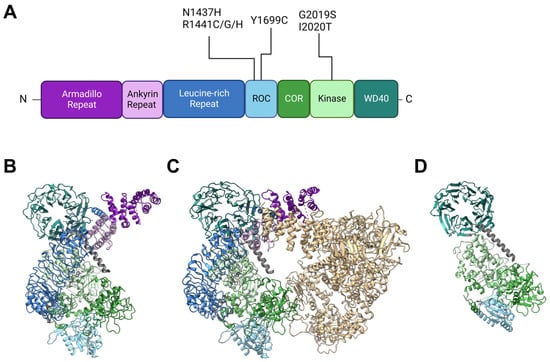
2. Insights from Genetic and Structural Studies of LRRK2
Back in 2002, Funayama et al. reported a family of inherited PD with an unknown causative gene locus, PARK8 [20][14]. Two years later, two independent groups identified the responsible gene as LRRK2 [1,2][1][2]. Since then, genetics have revealed at least seven causative mutations in this gene product: N1437H [21][15], R1441C [1[1][16],22], R1441G [22][16], R1441H [22][16], Y1699C [1], G2019S [23][17], I2020T [1], and numerous other rare variants with unclear causality [24][18]. The gene product LRRK2 is a 2527-amino-acid multidomain kinase, and the PD-associated mutations lie in the ROC (Ras of complex), COR (C-terminal of ROC), and kinase domains (Figure 1). The ROC domain is a GTP-binding domain that regulates the kinase activity in an intramolecular fashion [25[19][20],26], although its GTP-binding state (not GTP-binding capacity) may not be required for its kinase activity [27][21]. Other domains in LRRK2 have no PD-associated mutants allocated to them but are also of importance when considering intermolecular interactions. Armadillo repeats (ARM), ankyrin repeats (ANK), and leucine-rich repeats (LRR) are relatively abundant motifs that form various sizes of scaffolds for protein interaction [33[22][23][24],34,35], whereas WD40 domains are beta-propellers that may interact with DNA as well as proteins [36,37][25][26]. The detailed primary structure is depicted in Figure 1A. It might also be worth noting that a portion of endogenous LRRK2 in macrophages is found cleaved at the ANK-LRR interdomain region to produce a C-terminal fragment including the kinase region [38][27]. Although this fragment may be nonfunctional because the N-terminal membrane-interacting region is lacking, it may also act as dominant negative as it can heterodimerize with full-length LRRK2 [38][27].Figure 1. Structures of LRRK2 and PD-associated mutations. (A) Primary structure of LRRK2 and the location of reported PD-associated mutations. ROC: Ras of complex domain, COR: C terminal of ROC domain, WD40: WD40 domain. Domain lengths and borders are based on [39][28]. (B) Monomeric structure of full-length LRRK2 [30][29] (PDB ID: 7HLT). (C) Homodimeric structure of full-length LRRK2 [30][29] (PDB ID: 7HLW). (D) Protomer of filamentous LRRK2 C-terminal half (RCKW) on microtubules [40][30] (PDB ID: 6VNO). All structures are aligned so that the WD40 domain lies at the top left. Colorization of structures is the same for (A–D).
LRRK2 is observed in many conformations in vitro or in cells, with structural models from monomer [30][29] to homodimer [30,41,42][29][31][32] to filamentous [40,43][30][33] and even heteromultimer [44][34] reported (Figure 1B–D). The classical model of LRRK2 is the homodimer model, based on biochemical analysis and crystal structures of ROC domains [45,46][35][36]. This model was further confirmed via other methods such as cross-linking and negative stain [47][37], and further with full-length LRRK2 [30][29] (Figure 1B).
The monomeric model was first proposed based on biochemical analysis of full-length LRRK2 [50][38] but was not confirmed until some years ago when the structure of full-length LRRK2 was finally determined via cryoelectron microscopy (cryo-EM) [30][29] (Figure 1C).
The filamentous LRRK2 model is based on observations of overexpressed PD-associated mutants, which are mostly cytosolic but often form filaments along microtubules [51][39]. Reports that analyzed this form of LRRK2 bound to microtubules via cryo-EM or cryoelectron tomography (cryo-ET) showed multimer formation via interactions through the WD40 domain with the help of microtubule filaments [40,43][30][33] (a protomer of the filament is shown in Figure 1D).
3. Rab GTPases and LRRK2
3.1. Rab GTPases and the Endolysosomal System
Rab GTPases are proteins that form a subfamily of the Ras superfamily and are capable of binding cellular membranes via the C-terminal prenylation. Like other small GTPases, they switch their activity, which influences their affinity to specific proteins called effectors by changing their binding state with either GTP (active) or GDP (inactive) with the help of their specific guanine nucleotide exchange factors (GEF) or GTPase-activating protein (GAP). Their functions are associated with a wide variety of intracellular trafficking, ranging from cellular secretory pathways to intracellular degradation pathways involving the endolysosomal system. The endolysosomal system is part of an intracellular flow of enveloped membrane organelles and, apart from its function in autophagy, serves as a sorting site for substances incorporated by endocytosis. These substances include extracellular materials as well as membrane proteins and cellular membranes themselves. These substances are then either guided towards degradation by lysosomes, returned (or “recycled”) to the plasma membrane, or routed to the TGN. This pathway is known to regulate basic steps of cellular processes such as signaling, adhesion, immunity, nutrient uptake, organelle homeostasis, membrane protein turnover, and much more (reviewed in [57,58,59,60,61,62][40][41][42][43][44][45]). This route to degradation can be broken up into several parts, with more than one Rab GTPase regulating the routing or maturation of each vesicle. Endocytosed materials are first retained in the early endosome, where Rab5 controls the maturation of the vesicle and other Rab GTPases, such as Rab11, regulate re-routing from the early endosome to other compartments, in this case to the recycling endosomes, another part of the endolysosomal system, and ultimately to the plasma membrane [63,64][46][47].3.2. LRRK2 and Substrate Rab GTPases
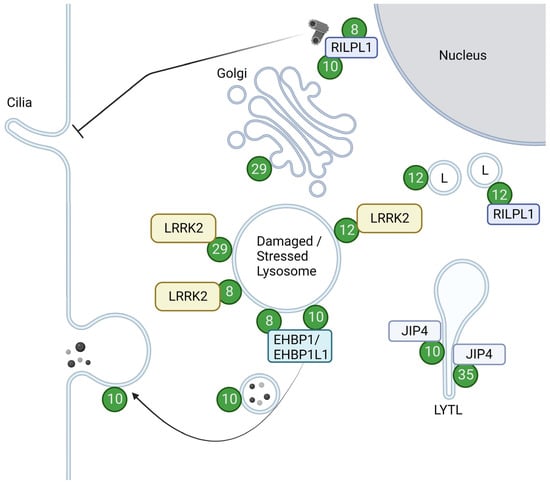
As a kinase, LRRK2 phosphorylates a subset of Rab GTPases, e.g., Rab8 and Rab10, in cells. The overview of the Rabs with their roles upon phosphorylation is summarized in Figure 2.Figure 2. Localizations and functions of LRRK2-phosphorylated Rabs. Green circles with numbers indicate Rabs phosphorylated by LRRK2. LRRK2-phosphorylated Rab12 and Rab29 are recruited to damaged or stressed lysosomes and further activate LRRK2. LRRK2-phosphorylated Rab8 and Rab10, bound to their effector RILPL1, accumulate near centrosomes to inhibit ciliogenesis. Rab8 and Rab10 also act together with their effector EHBP1 or EHBP1L1 to counteract lysosomal inflation or facilitate lysosomal release. LRRK2-phosphorylated Rab10 and Rab35 bind to JIP4 and induce LYTL. LRRK2-phosphorylated Rab12 binds to RILPL1 and moves lysosomes to the perinuclear region. LRRK2-phosphorylated Rab29 alters the morphology of the trans-Golgi. L: Lysosomes. Figure created with BioRender.com.
3.2.1. Rab8 and Rab10
Rab8 and Rab10 are closely related Rabs, both categorized in the Rab8 subfamily [89][48] and are the most characterized Rab GTPase in the context of interaction with LRRK2.
Right after the initial report of multiple Rab phosphorylation by LRRK2 [14][49], Rab10 was found to be a very sensitive marker for assessing LRRK2 activity [90][50], followed by a quick development of a phospho-specific antibody against Rab10 [68][51]. To date, numerous studies have incorporated an assessment of this phosphorylation in their studies on LRRK2 kinase activity [24,55,76,77,78,81,82,85,91,92,93,94,95,96,97,98,99][18][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67].
Rab10 has been implicated in several modes of transport in a variety of cell types, from general exocytic pathways to neurite or cilia formation and immune responses [110][68]. LRRK2 has been shown to play a role in these functions via its kinase activity, altering the ability of Rab10 to interact with other proteins. In the context of ciliogenesis, phosphorylated Rab10 binds to RILPL1 at or near centrosomes, inhibiting ciliogenesis [15,71,78,79,80][55][69][70][71][72]. This inhibition was also brought about by the retention of Myosin Va at centrioles by the same phosphorylation of Rab10 [77][54].
Rab8, on the other hand, is well characterized as a Rab GTPase, with GEFs Rabin8 and GRAB, GAPs TBC1D30, and other TBC family proteins, and multiple effectors described in the context of anterograde trafficking, endocytic recycling and exocytosis, association with the cytoskeleton, cell shape regulation and migration, ciliogenesis, neurite growth, and much more [111][73].
The first reports on Rab8 functions date back about 30 years when Rab8 was deemed responsible for post-Golgi anterograde trafficking in epithelial and neuronal cells [113,114][74][75]. Studies on Rab8’s involvement in neurite formation immediately followed [115][76] and built the classical view of Rab8 as a controller of neurite formation via anterograde trafficking. Current knowledge on Rab8 in neurite formation includes additional upstream elements, involving various other Rabs and their effectors, such as Rab11 and Rabin8, that activate Rab8 and Rab10 for neurite outgrowth [116][77], as well as downstream elements such as Cdc42 and tuba that strictly regulate the number of axons formed per cell [117][78].
Another aspect of Rab8 is its involvement in cilia. Formation of cilia requires Rab8 activation on the centrosome by Rabin8, very much like in neurite formation, and further protein trafficking to the base of primary cilia [111][73]. Impaired receptor trafficking to cilia via Rab8 dysfunction causes various deficits in functions that require cilia, such as adipocyte differentiation, where Rab8 is responsible for the trafficking of frz2 to the base of primary cilia [118][79]. Rab10 and Rab13 might have compensatory roles in ciliogenesis as double knockouts of Rab8a and Rab8b were insufficient to cause cilial deficits [119][80].
3.2.2. Rab29
Also known as Rab7L1, Rab29 itself is nominated as a risk factor for PD, encoded in the PARK16 locus [125,126,127][81][82][83]. This GTPase was among the first of all Rabs to have their relationship with LRRK2 uncovered [128,129][84][85] and is the Rab that is known to regulate LRRK2 from upstream [16,72,76,87,92,130][53][60][86][87][88][89] in addition to the recently reported Rab12 [53,54][90][91] and Rab38 [131][92]. Rab29 localization at steady state was reported to be at the Golgi, with a small fraction at perinuclear vesicles [132,133][93][94]. These Rab29 population at the Golgi was reported to be responsible for the integrity of the TGN and the retrograde trafficking there [18,134][95][96]. Golgi fragmentation was also dependent on the recruitment and activation of LRRK2 induced by Rab29 overexpression [16[86][97],19], as well as phosphorylation of Rab29 by LRRK2 [18][95]. The function of Rab29 has attracted attention not only from its relationship to the Golgi but also to the lysosome, as the small population of Rab29 at perinuclear vesicles was found to be lysosomal. Also, Rab29 has been shown to react to lysosomal stress, localizing itself to lysosomes and also co-recruiting and activating LRRK2 [76,87][53][88]. Active Rab29 on lysosomes regulates the size of abnormally inflated lysosomes, which is dependent on LRRK2 kinase activity [76,87][53][88].3.2.3. Rab12
Rab12 was first found to regulate a “non-canonical” degradation route from recycling endosomes directly to lysosomes [136][98], then further allocated to more transfer between the cell surface and Golgi for various cargoes [137,138][99][100]. Rab12 is also gradually being understood as a potent marker of LRRK2 activity, as the phosphorylation of Rab12 was reported to be potently induced by PD-associated mutants of LRRK2 [133,139][94][101]. The functions of the phosphorylation of this GTPase were not known until very recently; it was found to be responsible for controlling the intracellular localization of lysosomes via an increase in the binding ability to RILPL1 [86][102]. An unusual point about this phosphorylation is that LRRK2 recognizes GDP-bound Rab12 better than the GTP-bound form, at least in vitro [140][103]. Rab12 is also implicated in lysosomal repair, as Rab12 accumulates on damaged lysosomes and activates LRRK2 there [53,54][90][91]. Rab12-mediated accumulation and activation of LRRK2 on lysosomes during lysosomal damage were enhanced in PD-associated mutants of LRRK2 or even VPS35, even under non-damaged conditions, but were not enhanced beyond wild-type during damage [54][91], suggestive of a Rab12-dependent lysosomal response mechanism that might be constantly activated in the course of PD pathogenesis with these mutations.3.2.4. Rab35
Rab35 is a Rab GTPase responsible for various cellular processes including exosome release, neurite outgrowth, phagocytosis, cell polarization, immune synapse formation, cytokinesis, and cell migration [141][104]. These pathways are controlled by either the quick recycling of endocytic cargoes (e.g., T cell receptor (TCR) and MHC complexes for immune synapse formation, podocalyxin for cell polarization) to the plasma membrane or the regulation of actin beneath the plasma membrane to promote changes in cell shape and position [141][104]. Although the main link to diseases would be between cancer, there are several reports that link this GTPase and LRRK2 to PD. The effectors of Rab35 include OCRL, MICAL1, and MICAL-L1 [132][93], which are also effectors of Rab8 or Rab10 as noted above. Not surprisingly, some of the functions of Rab35 overlap with Rab8 and Rab10, which include the recruitment of JIP4 and induction of LYTL upon phosphorylation by LRRK2 [83][105]. LRRK2-induced phosphorylation of Rab35 was also found to positively regulate the propagation of α-synuclein [88][106].3.2.5. Rab5
Rab5 is the key GTPase in controlling the maturation of early endosomes. After endocytosis, Rab5 is recruited to the endocytic vesicle by Rab4 or the Rab5 GEF Rabex5 (RABGEF1), and then in turn recruits effector proteins such as EEA1, a tether for fusion with other early endosomes, VPS34, a phosphatidylinositol kinase responsible for converting phosphatidylinositol (PI) to the endosome-enriched lipid phosphatidylinositol-3-phosphate (PI3P), and the Rab7-GEF Mon1-Ccz1, which recruits Rab7 and facilitates transition to late endosomes [60][43]. Although there are many studies on Rab5, the differences between the three isoforms Rab5a, Rab5b, and Rab5c are not that undeciphered. Rab5c is reported to have a slightly different function from the other two, with little involvement in EGFR recycling [142][107] or specific involvement in Rac1-dependent cell migration [143][108]. Rab5 and PD have little connection reported, with implications in Rab5a-mediated uptake of α-synuclein in neurons [145][109] or clearance in microglia [146][110]. The latter is the phenotype also seen in LRRK2 knockout mice, which could hint at the possibility of Rab5 interplay in PD pathogenesis or treatment. Other links reported between LRRK2 and Rab5 include a cooperative regulation of neurite outgrowth [147][111], phosphorylation of all the isoforms of Rab5 by LRRK2 [15][69], and inactivation of Rab5b [70][112].3.2.6. Rab3
Rab3 has four isoforms (Rab3a, Rab3b, Rab3c, Rab3d), and all of the isoforms participate in exocytosis or secretion. They are highly expressed in neurons and secretory cells [66,148][113][114]. Their roles in secretion in secretory cells or neurons appear to be redundant, with several knockout studies in mice observing little or no changes in exocytotic activity, but depletion of all Rab3 isoforms results in lethality from respiratory failure [149,150,151,152][115][116][117][118]. In neurons, Rab3 regulates a specific type of exocytotic vesicles called dense-core vesicles, which are important in neuropeptide release [153][119]. The four isoforms display different magnitudes of activity, with Rab3a being the most active [153][119]. Rab3a is also found to be responsible for plasma membrane repair via lysosomal exocytosis [154][120]. Although Rab3 was identified as a LRRK2 substrate, there is only a limited number of studies on the interaction between these two proteins. One shows that Rab3a colocalizes with LRRK2 on stressed lysosomes dependent on its kinase activity [76][53], and another shows that Rab3 is a very weak substrate in LRRK2-G2019S expressing neurons, probably because of different localizations in neurons [159][121].3.2.7. Rab1
Rab1 is the newest Rab GTPase found to be a substrate of LRRK2 [69][122]. The classical role of Rab1 is its involvement in ER-Golgi transport and maintenance of the Golgi, but its functions reach out to regulating the localization of endosomes and lysosomes, and consequential cell-surface receptor recycling [160][123]. Loss of Rab1 results in fragmentation of the Golgi, which is seen in α-synuclein overexpression models [13] or in dopaminergic neurons in the substantia nigra of PD patients [161][124].4. Rab Phosphorylation in Relation to PD
As stated above, almost all familial PD mutations increase Rab phosphorylation in cells, although the most common G2019S mutation may have somewhat different effects than others. That is, the G2019S mutation increases its intrinsic kinase activity more effectively than Rab phosphorylation, whereas the other mutations increase Rab phosphorylation more potently than its kinase activity [24][18]. This is consistent with the observations in human samples; the increase in Rab10 phosphorylation has been shown in peripheral blood neutrophils of R1441G mutation carriers [106][125], whereas G2019S mutation appears to have a weaker effect on Rab10 phosphorylation induction, at least in neutrophils [106][125] and peripheral blood mononuclear cells (PBMCs) [105,108][126][127]. These observations implicate slightly different pathomechanisms of PD for G2019S and other familial mutations. In relevance to the pathomechanism of PD, one should take into account specific cell types in the brain, such as neurons and glia, as LRRK2 and each Rab are known to be expressed relatively ubiquitously in these cells. In analyses using mouse primary neurons and glia, Rab10 phosphorylation is detected in all cell types but is more strongly detected in astrocytes and microglia [105][126]. In vivo, it has been shown that cholinergic neurons in the striatum of LRRK2 R1441C knock-in mice develop ciliation defects, likely due to Rab10 over-phosphorylation [79][71], and a similar ciliation phenotype was subsequently observed also in astrocytes of LRRK2 G2019S knock-in mice [166][128].5. Conclusions
From a cell biology viewpoint, it would be interesting to further clarify what Rab over-phosphorylation causes in cells. As only a small fraction of Rabs is phosphorylated at steady state, it is likely that the majority of a given pool of Rab proteins can still carry out their normal functions [90][50]. On the other hand, phosphorylated Rab8a and Rab10 have been shown to bind new effector proteins, i.e., RILPL1/2 and JIP3/4 [15,74[69][105][129],83], suggesting that even a small fraction of them may dominantly affect cellular functions. Indeed, it has been shown that the recruitment of RILPL1 regulates ciliogenesis and centrosomal cohesion [15,71][69][70] and that of JIP4 regulates lysosomal tubulation [83][105] and axonal autophagosome transport [170][130]. In addition, as noted above, Rab10 phosphorylation by LRRK2 is markedly enhanced under lysosomal stress and is therefore assumed to play important roles in the maintenance of endolysosomes. The effects on disease-causing aggregate-prone proteins are also of interest; the brains of patients with LRRK2 mutations often, but not always, accumulate insoluble α-synuclein and tau to varying degrees, leading to the notion that Rab over-phosphorylation may also affect the metabolism or propagation of these proteins. Indeed, as mentioned above, phosphorylation of Rab35 has been shown to potentially regulate α-synuclein propagation [88][106]. With respect to clinical applications, LRRK2 inhibitors including small molecule compounds and antisense oligonucleotides are being developed, and one of them, BIIB122/DNL151, originally developed by Denali Therapeutics Inc, has now proceeded to phase III trials [171][131]. According to their clinical trial reports, administration of this LRRK2 inhibitor has been shown to markedly reduce Rab10 phosphorylation in PBMCs, as well as phospho-Ser935 LRRK2 in whole blood, total LRRK2 in cerebrospinal fluid (CSF), and a lysosomal lipid di-22:6-bis (monoacylglycerol) phosphate (BMP) in urine, all in a dose-dependent manner [172][132]. Also, the clinical trial results of another LRRK2 inhibitor, DNL201, were reported earlier, and the results were mostly similar [107][133]. IReferences
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607.
- Paisán-Ruíz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simón, J.; van der Brug, M.; de Munain, A.L.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the Gene Containing Mutations That Cause PARK8-Linked Parkinson’s Disease. Neuron 2004, 44, 595–600.
- Manzoni, C.; Lewis, P.A. Leucine-Rich Repeat Kinase 2 (LRRK2). Adv. Neurobiol. 2017, 14, 89–105.
- Singh, F.; Ganley, I.G. Parkinson’s Disease and Mitophagy: An Emerging Role for LRRK2. Biochem. Soc. Trans. 2021, 49, 551–562.
- Berwick, D.C.; Heaton, G.R.; Azeggagh, S.; Harvey, K. LRRK2 Biology from Structure to Dysfunction: Research Progresses, but the Themes Remain the Same. Mol. Neurodegener. 2019, 14, 49.
- Boecker, C.A. The Role of LRRK2 in Intracellular Organelle Dynamics. J. Mol. Biol. 2023, 435, 167998.
- Herbst, S.; Gutierrez, M.G. LRRK2 in Infection: Friend or Foe? ACS Infect. Dis. 2019, 5, 809–815.
- Kuwahara, T.; Iwatsubo, T. The Emerging Functions of LRRK2 and Rab GTPases in the Endolysosomal System. Front. Neurosci. 2020, 14, 227.
- Taylor, M.; Alessi, D.R. Advances in Elucidating the Function of Leucine-Rich Repeat Protein Kinase-2 in Normal Cells and Parkinson’s Disease. Curr. Opin. Cell Biol. 2020, 63, 102–113.
- Erb, M.L.; Moore, D.J. LRRK2 and the Endolysosomal System in Parkinson’s Disease. J. Park. Dis. 2020, 10, 1271–1291.
- Norris, A.; Grant, B.D. Endosomal Microdomains: Formation and Function. Curr. Opin. Cell Biol. 2020, 65, 86–95.
- Dalfó, E.; Gómez-Isla, T.; Rosa, J.L.; Bodelón, M.N.; Tejedor, M.C.; Barrachina, M.; Ambrosio, S.; Ferrer, I. Abnormal α-Synuclein Interactions with Rab Proteins in α-Synuclein A30P Transgenic Mice. J. Neuropathol. Exp. Neurol. 2004, 63, 302–313.
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. α-Synuclein Blocks ER-Golgi Traffic and Rab1 Rescues Neuron Loss in Parkinson’s Models. Science 2006, 313, 324–328.
- Funayama, M.; Hasegawa, K.; Kowa, H.; Saito, M.; Tsuji, S.; Obata, F. A New Locus for Parkinson’s Disease (PARK8) Maps to Chromosome 12p11.2–Q13.1. Ann. Neurol. 2002, 51, 296–301.
- Aasly, J.O.; Vilariño-Güell, C.; Dachsel, J.C.; Webber, P.J.; West, A.B.; Haugarvoll, K.; Johansen, K.K.; Toft, M.; Nutt, J.G.; Payami, H.; et al. Novel Pathogenic LRRK2 p.Asn1437His Substitution in Familial Parkinson’s Disease. Mov. Disord. 2010, 25, 2156–2163.
- Mata, I.F.; Kachergus, J.M.; Taylor, J.P.; Lincoln, S.; Aasly, J.; Lynch, T.; Hulihan, M.M.; Cobb, S.A.; Wu, R.-M.; Lu, C.-S.; et al. Lrrk2 Pathogenic Substitutions in Parkinson’s Disease. Neurogenetics 2005, 6, 171–177.
- Kachergus, J.; Mata, I.F.; Hulihan, M.; Taylor, J.P.; Lincoln, S.; Aasly, J.; Gibson, J.M.; Ross, O.A.; Lynch, T.; Wiley, J.; et al. Identification of a Novel LRRK2 Mutation Linked to Autosomal Dominant Parkinsonism: Evidence of a Common Founder across European Populations. Am. J. Hum. Genet. 2005, 76, 672–680.
- Kalogeropulou, A.F.; Purlyte, E.; Tonelli, F.; Lange, S.M.; Wightman, M.; Prescott, A.R.; Padmanabhan, S.; Sammler, E.; Alessi, D.R. Impact of 100 LRRK2 Variants Linked to Parkinson’s Disease on Kinase Activity and Microtubule Binding. Biochem. J. 2022, 479, 1759–1783.
- Bosgraaf, L.; Haastert, P.J.M.V. Roc, a Ras/GTPase Domain in Complex Proteins. Biochim. Biophys. Acta BBA Mol. Cell Res. 2003, 1643, 5–10.
- Guo, L.; Gandhi, P.N.; Wang, W.; Petersen, R.B.; Wilson-Delfosse, A.L.; Chen, S.G. The Parkinson’s Disease-Associated Protein, Leucine-Rich Repeat Kinase 2 (LRRK2), Is an Authentic GTPase Thatstimulates Kinase Activity. Exp. Cell Res. 2007, 313, 3658–3670.
- Taymans, J.-M.; Vancraenenbroeck, R.; Ollikainen, P.; Beilina, A.; Lobbestael, E.; Maeyer, M.D.; Baekelandt, V.; Cookson, M.R. LRRK2 Kinase Activity Is Dependent on LRRK2 GTP Binding Capacity but Independent of LRRK2 GTP Binding. PLoS ONE 2011, 6, e23207.
- Mosavi, L.K.; Cammett, T.J.; Desrosiers, D.C.; Peng, Z. The Ankyrin Repeat as Molecular Architecture for Protein Recognition. Protein Sci. 2004, 13, 1435–1448.
- Tewari, R.; Bailes, E.; Bunting, K.A.; Coates, J.C. Armadillo-Repeat Protein Functions: Questions for Little Creatures. Trends Cell Biol. 2010, 20, 470–481.
- Matsushima, N.; Takatsuka, S.; Miyashita, H.; Kretsinger, R.H. Leucine Rich Repeat Proteins: Sequences, Mutations, Structures and Diseases. Protein Pept. Lett. 2019, 26, 108–131.
- Xu, C.; Min, J. Structure and Function of WD40 Domain Proteins. Protein Cell 2011, 2, 202–214.
- Schapira, M.; Tyers, M.; Torrent, M.; Arrowsmith, C.H. WD40 Repeat Domain Proteins: A Novel Target Class? Nat. Rev. Drug Discov. 2017, 16, 773–786.
- Lee, H.; Flynn, R.; Sharma, I.; Haberman, E.; Carling, P.J.; Nicholls, F.J.; Stegmann, M.; Vowles, J.; Haenseler, W.; Wade-Martins, R.; et al. LRRK2 Is Recruited to Phagosomes and Co-Recruits RAB8 and RAB10 in Human Pluripotent Stem Cell-Derived Macrophages. Stem Cell Rep. 2020, 14, 940–955.
- Ito, G.; Utsunomiya-Tate, N. Overview of the Impact of Pathogenic LRRK2 Mutations in Parkinson’s Disease. Biomolecules 2023, 13, 845.
- Myasnikov, A.; Zhu, H.; Hixson, P.; Xie, B.; Yu, K.; Pitre, A.; Peng, J.; Sun, J. Structural Analysis of the Full-Length Human LRRK2. Cell 2021, 184, 3519–3527.e10.
- Deniston, C.K.; Salogiannis, J.; Mathea, S.; Snead, D.M.; Lahiri, I.; Matyszewski, M.; Donosa, O.; Watanabe, R.; Böhning, J.; Shiau, A.K.; et al. Structure of LRRK2 in Parkinson’s Disease and Model for Microtubule Interaction. Nature 2020, 588, 344–349.
- Guaitoli, G.; Gilsbach, B.K.; Raimondi, F.; Gloeckner, C.J. First Model of Dimeric LRRK2: The Challenge of Unrevealing the Structure of a Multidomain Parkinson’s-Associated Protein. Biochem. Soc. Trans. 2016, 44, 1635–1641.
- Zhang, P.; Fan, Y.; Ru, H.; Wang, L.; Magupalli, V.G.; Taylor, S.S.; Alessi, D.R.; Wu, H. Crystal Structure of the WD40 Domain Dimer of LRRK2. Proc. Natl. Acad. Sci. USA 2019, 116, 1579–1584.
- Watanabe, R.; Buschauer, R.; Böhning, J.; Audagnotto, M.; Lasker, K.; Lu, T.-W.; Boassa, D.; Taylor, S.; Villa, E. The In Situ Structure of Parkinson’s Disease-Linked LRRK2. Cell 2020, 182, 1508–1518.e16.
- Zhu, H.; Tonelli, F.; Alessi, D.R.; Sun, J. Structural Basis of Human LRRK2 Membrane Recruitment and Activation. bioRxiv 2022.
- Greggio, E.; Zambrano, I.; Kaganovich, A.; Beilina, A.; Taymans, J.-M.; Daniëls, V.; Lewis, P.; Jain, S.; Ding, J.; Syed, A.; et al. The Parkinson Disease-Associated Leucine-Rich Repeat Kinase 2 (LRRK2) Is a Dimer That Undergoes Intramolecular Autophosphorylation*. J. Biol. Chem. 2008, 283, 16906–16914.
- Deng, J.; Lewis, P.A.; Greggio, E.; Sluch, E.; Beilina, A.; Cookson, M.R. Structure of the ROC Domain from the Parkinson’s Disease-Associated Leucine-Rich Repeat Kinase 2 Reveals a Dimeric GTPase. Proc. Natl. Acad. Sci. USA 2008, 105, 1499–1504.
- Guaitoli, G.; Raimondi, F.; Gilsbach, B.K.; Gómez-Llorente, Y.; Deyaert, E.; Renzi, F.; Li, X.; Schaffner, A.; Jagtap, P.K.A.; Boldt, K.; et al. Structural Model of the Dimeric Parkinson’s Protein LRRK2 Reveals a Compact Architecture Involving Distant Interdomain Contacts. Proc. Natl. Acad. Sci. USA 2016, 113, E4357–E4366.
- Ito, G.; Iwatsubo, T. Re-Examination of the Dimerization State of Leucine-Rich Repeat Kinase 2: Predominance of the Monomeric Form. Biochem. J. 2012, 441, 987–998.
- Kett, L.R.; Boassa, D.; Ho, C.C.-Y.; Rideout, H.J.; Hu, J.; Terada, M.; Ellisman, M.; Dauer, W.T. LRRK2 Parkinson Disease Mutations Enhance Its Microtubule Association. Hum. Mol. Genet. 2012, 21, 890–899.
- Cullen, P.J.; Steinberg, F. To Degrade or Not to Degrade: Mechanisms and Significance of Endocytic Recycling. Nat. Rev. Mol. Cell Biol. 2018, 19, 679–696.
- Scott, C.C.; Vacca, F.; Gruenberg, J. Endosome Maturation, Transport and Functions. Semin. Cell Dev. Biol. 2014, 31, 2–10.
- Wang, J.; Fedoseienko, A.; Chen, B.; Burstein, E.; Jia, D.; Billadeau, D.D. Endosomal Receptor Trafficking: Retromer and Beyond. Traffic 2018, 19, 578–590.
- Langemeyer, L.; Fröhlich, F.; Ungermann, C. Rab GTPase Function in Endosome and Lysosome Biogenesis. Trends Cell Biol. 2018, 28, 957–970.
- Gruenberg, J. Life in the Lumen: The Multivesicular Endosome. Traffic 2020, 21, 76–93.
- O’Sullivan, M.J.; Lindsay, A.J. The Endosomal Recycling Pathway—At the Crossroads of the Cell. Int. J. Mol. Sci. 2020, 21, 6074.
- Huotari, J.; Helenius, A. Endosome Maturation. EMBO J. 2011, 30, 3481–3500.
- Campa, C.C.; Margaria, J.P.; Derle, A.; Giudice, M.D.; Santis, M.C.D.; Gozzelino, L.; Copperi, F.; Bosia, C.; Hirsch, E. Rab11 Activity and PtdIns(3)P Turnover Removes Recycling Cargo from Endosomes. Nat. Chem. Biol. 2018, 14, 801–810.
- Homma, Y.; Hiragi, S.; Fukuda, M. Rab Family of Small GTPases: An Updated View on Their Regulation and Functions. FEBS J. 2021, 288, 36–55.
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics Reveals That Parkinson’s Disease Kinase LRRK2 Regulates a Subset of Rab GTPases. eLife 2016, 5, e12813.
- Ito, G.; Katsemonova, K.; Tonelli, F.; Lis, P.; Baptista, M.; Shpiro, N.; Duddy, G.; Wilson, S.; Ho, P.; Ho, S.-L.; et al. Phos-Tag Analysis of Rab10 Phosphorylation by LRRK2: A Powerful Assay for Assessing Kinase Function and Inhibitors. Biochem. J. 2016, 473, 2671–2685.
- Lis, P.; Burel, S.; Steger, M.; Mann, M.; Brown, F.; Diez, F.; Tonelli, F.; Holton, J.L.; Ho, P.W.; Ho, S.-L.; et al. Development of Phospho-Specific Rab Protein Antibodies to Monitor in Vivo Activity of the LRRK2 Parkinson’s Disease Kinase. Biochem. J. 2018, 475, BCJ20170802.
- Kuwahara, T.; Funakawa, K.; Komori, T.; Sakurai, M.; Yoshii, G.; Eguchi, T.; Fukuda, M.; Iwatsubo, T. Roles of Lysosomotropic Agents on LRRK2 Activation and Rab10 Phosphorylation. Neurobiol. Dis. 2020, 145, 105081.
- Eguchi, T.; Kuwahara, T.; Sakurai, M.; Komori, T.; Fujimoto, T.; Ito, G.; Yoshimura, S.; Harada, A.; Fukuda, M.; Koike, M.; et al. LRRK2 and Its Substrate Rab GTPases Are Sequentially Targeted onto Stressed Lysosomes and Maintain Their Homeostasis. Proc. Natl. Acad. Sci. USA 2018, 115, E9115–E9124.
- Dhekne, H.S.; Yanatori, I.; Vides, E.G.; Sobu, Y.; Diez, F.; Tonelli, F.; Pfeffer, S.R. LRRK2-Phosphorylated Rab10 Sequesters Myosin Va with RILPL2 during Ciliogenesis Blockade. Life Sci. Alliance 2021, 4, e202101050.
- Ordóñez, A.J.L.; Fasiczka, R.; Fernández, B.; Naaldijk, Y.; Fdez, E.; Ramírez, M.B.; Phan, S.; Boassa, D.; Hilfiker, S. The LRRK2 Signaling Network Converges on a Centriolar Phospho-Rab10/RILPL1 Complex to Cause Deficits in Centrosome Cohesion and Cell Polarization. Biol. Open 2022, 11, bio059468.
- Liu, Z.; Xu, E.; Zhao, H.T.; Cole, T.; West, A.B. LRRK2 and Rab10 Coordinate Macropinocytosis to Mediate Immunological Responses in Phagocytes. EMBO J. 2020, 39, e104862.
- Kluss, J.H.; Beilina, A.; Williamson, C.D.; Lewis, P.A.; Cookson, M.R.; Bonet-Ponce, L. Lysosomal Positioning Regulates Rab10 Phosphorylation at LRRK2+ Lysosomes. Proc. Natl. Acad. Sci. USA 2022, 119, e2205492119.
- Li, J.; Wu, M.; Gong, Y.; Tang, J.; Shen, J.; Xu, L.; Dang, B.; Chen, G. Inhibition of LRRK2-Rab10 Pathway Improves Secondary Brain Injury After Surgical Brain Injury in Rats. Front. Surg. 2022, 8, 749310.
- Fan, Y.; Howden, A.J.; Sarhan, A.R.; Lis, P.; Ito, G.; Martinez, T.N.; Brockmann, K.; Gasser, T.; Alessi, D.R.; Sammler, E.M. Interrogating Parkinson’s Disease LRRK2 Kinase Pathway Activity by Assessing Rab10 Phosphorylation in Human Neutrophils. Biochem. J. 2017, 475, 23–44.
- Gomez, R.C.; Wawro, P.; Lis, P.; Alessi, D.R.; Pfeffer, S.R. Membrane Association but Not Identity Is Required for LRRK2 Activation and Phosphorylation of Rab GTPases. J. Cell Biol. 2019, 218, 4157–4170.
- Korecka, J.A.; Thomas, R.; Hinrich, A.J.; Moskites, A.M.; Macbain, Z.K.; Hallett, P.J.; Isacson, O.; Hastings, M.L. Splice-Switching Antisense Oligonucleotides Reduce LRRK2 Kinase Activity in Human LRRK2 Transgenic Mice. Mol. Ther. Nucleic Acids 2020, 21, 623–635.
- Iannotta, L.; Biosa, A.; Kluss, J.H.; Tombesi, G.; Kaganovich, A.; Cogo, S.; Plotegher, N.; Civiero, L.; Lobbestael, E.; Baekelandt, V.; et al. Divergent Effects of G2019S and R1441C LRRK2 Mutations on LRRK2 and Rab10 Phosphorylations in Mouse Tissues. Cells 2020, 9, 2344.
- Kelly, K.; Chang, A.; Hastings, L.; Abdelmotilib, H.; West, A.B. Genetic Background Influences LRRK2-Mediated Rab Phosphorylation in the Rat Brain. Brain Res. 2021, 1759, 147372.
- Nazish, I.; Arber, C.; Piers, T.M.; Warner, T.T.; Hardy, J.A.; Lewis, P.A.; Pocock, J.M.; Bandopadhyay, R. Abrogation of LRRK2 Dependent Rab10 Phosphorylation with TLR4 Activation and Alterations in Evoked Cytokine Release in Immune Cells. Neurochem. Int. 2021, 147, 105070.
- Keeney, M.; Hoffman, E.; Greenamyre, J.; Maio, R.D. Measurement of LRRK2 Kinase Activity by Proximity Ligation Assay. Bio-protocol 2021, 11, e4140.
- Tasegian, A.; Singh, F.; Ganley, I.G.; Reith, A.D.; Alessi, D.R. Impact of Type II LRRK2 Inhibitors on Signaling and Mitophagy. Biochem. J. 2021, 478, 3555–3573.
- Kluss, J.H.; Bonet-Ponce, L.; Lewis, P.A.; Cookson, M.R. Directing LRRK2 to Membranes of the Endolysosomal Pathway Triggers RAB Phosphorylation and JIP4 Recruitment. Neurobiol. Dis. 2022, 170, 105769.
- Chua, C.E.L.; Tang, B.L. Rab 10—A Traffic Controller in Multiple Cellular Pathways and Locations. J. Cell. Physiol. 2018, 233, 6483–6494.
- Steger, M.; Diez, F.; Dhekne, H.S.; Lis, P.; Nirujogi, R.S.; Karayel, O.; Tonelli, F.; Martinez, T.N.; Lorentzen, E.; Pfeffer, S.R.; et al. Systematic Proteomic Analysis of LRRK2-Mediated Rab GTPase Phosphorylation Establishes a Connection to Ciliogenesis. eLife 2017, 6, e31012.
- Ordóñez, A.; Fernández, B.; Fdez, E.; Romo-Lozano, M.; Madero-Pérez, J.; Lobbestael, E.; Baekelandt, V.; Aiastui, A.; Munaín, A.; Melrose, H.L.; et al. RAB8, RAB10 and RILPL1 Contribute to Both LRRK2 Kinase-Mediated Centrosomal Cohesion and Ciliogenesis Deficits. Hum. Mol. Genet. 2019, 28, 3552–3568.
- Dhekne, H.S.; Yanatori, I.; Gomez, R.C.; Tonelli, F.; Diez, F.; Schüle, B.; Steger, M.; Alessi, D.R.; Pfeffer, S.R. A Pathway for Parkinson’s Disease LRRK2 Kinase to Block Primary Cilia and Sonic Hedgehog Signaling in the Brain. eLife 2018, 7, e40202.
- Sobu, Y.; Wawro, P.S.; Dhekne, H.S.; Yeshaw, W.M.; Pfeffer, S.R. Pathogenic LRRK2 Regulates Ciliation Probability Upstream of Tau Tubulin Kinase 2 via Rab10 and RILPL1 Proteins. Proc. Natl. Acad. Sci. USA 2021, 118, e2005894118.
- Peränen, J. Rab8 GTPase as a Regulator of Cell Shape. Cytoskeleton 2011, 68, 527–539.
- Huber, L.A.; Pimplikar, S.; Parton, R.G.; Virta, H.; Zerial, M.; Simons, K. Rab8, a Small GTPase Involved in Vesicular Traffic between the TGN and the Basolateral Plasma Membrane. J. Cell Biol. 1993, 123, 35–45.
- Huber, L.A.; de Hoop, M.J.; Dupree, P.; Zerial, M.; Simons, K.; Dotti, C. Protein Transport to the Dendritic Plasma Membrane of Cultured Neurons Is Regulated by Rab8p. J. Cell Biol. 1993, 123, 47–55.
- Huber, L.A.; Dupree, P.; Dotti, C.G. A Deficiency of the Small GTPase Rab8 Inhibits Membrane Traffic in Developing Neurons. Mol. Cell. Biol. 1995, 15, 918–924.
- Homma, Y.; Fukuda, M. Rabin8 Regulates Neurite Outgrowth in Both GEF Activity–Dependent and –Independent Manners. Mol. Biol. Cell. 2016, 27, 2107–2118.
- Urrutia, P.J.; Bodaleo, F.; Bórquez, D.A.; Homma, Y.; Rozes-Salvador, V.; Villablanca, C.; Conde, C.; Fukuda, M.; González-Billault, C. Tuba Activates Cdc42 during Neuronal Polarization Downstream of the Small GTPase Rab8a. J. Neurosci. 2021, 41, 1636–1649.
- Stypulkowski, E.; Feng, Q.; Joseph, I.; Farrell, V.; Flores, J.; Yu, S.; Sakamori, R.; Sun, J.; Bandyopadhyay, S.; Das, S.; et al. Rab8 Attenuates Wnt Signaling and Is Required for Mesenchymal Differentiation into Adipocytes. J. Biol. Chem. 2021, 296, 100488.
- Sato, T.; Iwano, T.; Kunii, M.; Matsuda, S.; Mizuguchi, R.; Jung, Y.; Hagiwara, H.; Yoshihara, Y.; Yuzaki, M.; Harada, R.; et al. Rab8a and Rab8b Are Essential for Several Apical Transport Pathways but Insufficient for Ciliogenesis. J. Cell Sci. 2013, 127, 422–431.
- Simón-Sánchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-Wide Association Study Reveals Genetic Risk Underlying Parkinson’s Disease. Nat. Genet. 2009, 41, 1308–1312.
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-Wide Association Study Identifies Common Variants at Four Loci as Genetic Risk Factors for Parkinson’s Disease. Nat. Genet. 2009, 41, 1303–1307.
- Pihlstrøm, L.; Rengmark, A.; Bjørnarå, K.A.; Dizdar, N.; Fardell, C.; Forsgren, L.; Holmberg, B.; Larsen, J.P.; Linder, J.; Nissbrandt, H.; et al. Fine Mapping and Resequencing of the PARK16 Locus in Parkinson’s Disease. J. Hum. Genet. 2015, 60, 357–362.
- MacLeod, D.A.; Rhinn, H.; Kuwahara, T.; Zolin, A.; Di Paolo, G.; McCabe, B.D.; Marder, K.S.; Honig, L.S.; Clark, L.N.; Small, S.A.; et al. RAB7L1 Interacts with LRRK2 to Modify Intraneuronal Protein Sorting and Parkinson’s Disease Risk. Neuron 2013, 77, 425–439.
- Beilina, A.; Rudenko, I.N.; Kaganovich, A.; Civiero, L.; Chau, H.; Kalia, S.K.; Kalia, L.V.; Lobbestael, E.; Chia, R.; Ndukwe, K.; et al. Unbiased Screen for Interactors of Leucine-Rich Repeat Kinase 2 Supports a Common Pathway for Sporadic and Familial Parkinson Disease. Proc. Natl. Acad. Sci. USA 2014, 111, 2626–2631.
- Liu, Z.; Bryant, N.; Kumaran, R.; Beilina, A.; Abeliovich, A.; Cookson, M.R.; West, A.B. LRRK2 Phosphorylates Membrane-Bound Rabs and Is Activated by GTP-Bound Rab7L1 to Promote Recruitment to the Trans Golgi Network. Hum. Mol. Genet. 2018, 27, 385–395.
- Madero-Pérez, J.; Fernández, B.; Ordóñez, A.J.L.; Fdez, E.; Lobbestael, E.; Baekelandt, V.; Hilfiker, S. RAB7L1-Mediated Relocalization of LRRK2 to the Golgi Complex Causes Centrosomal Deficits via RAB8A. Front. Mol. Neurosci. 2018, 11, 417.
- Komori, T.; Kuwahara, T.; Fujimoto, T.; Sakurai, M.; Koyama-Honda, I.; Fukuda, M.; Iwatsubo, T. Phosphorylation of Rab29 at Ser185 Regulates Its Localization and Role in the Lysosomal Stress Response in Concert with LRRK2. J. Cell Sci. 2023, 136, jcs261003.
- Kuwahara, T.; Inoue, K.; D’Agati, V.D.; Fujimoto, T.; Eguchi, T.; Saha, S.; Wolozin, B.; Iwatsubo, T.; Abeliovich, A. LRRK2 and RAB7L1 Coordinately Regulate Axonal Morphology and Lysosome Integrity in Diverse Cellular Contexts. Sci. Rep. 2016, 6, 29945.
- Dhekne, H.S.; Tonelli, F.; Yeshaw, W.M.; Chiang, C.Y.; Limouse, C.; Jaimon, E.; Purlyte, E.; Alessi, D.R.; Pfeffer, S.R. Genome-Wide Screen Reveals Rab12 GTPase as a Critical Activator of Parkinson’s Disease-Linked LRRK2 Kinase. eLife 2023, 12, e87098.
- Wang, X.; Bondar, V.V.; Davis, O.B.; Maloney, M.T.; Agam, M.; Chin, M.Y.; Ho, A.C.-N.; Ghosh, R.; Leto, D.E.; Joy, D.; et al. Rab12 Is a Regulator of LRRK2 and Its Activation by Damaged Lysosomes. eLife 2023, 12, e87255.
- Unapanta, A.; Shavarebi, F.; Porath, J.; Shen, Y.; Balen, C.; Nguyen, A.; Tseng, J.; Leong, W.S.; Liu, M.; Lis, P.; et al. Endogenous Rab38 Regulates LRRK2’s Membrane Recruitment and Substrate Rab Phosphorylation in Melanocytes. J. Biol. Chem. 2023, 299, 105192.
- Helip-Wooley, A.; Thoene, J.G. Sucrose-Induced Vacuolation Results in Increased Expression of Cholesterol Biosynthesis and Lysosomal Genes. Exp. Cell Res. 2004, 292, 89–100.
- Kalogeropulou, A.F.; Freemantle, J.B.; Lis, P.; Vides, E.G.; Polinski, N.K.; Alessi, D.R. Endogenous Rab29 Does Not Impact Basal or Stimulated LRRK2 Pathway Activity. Biochem. J. 2020, 477, 4397–4423.
- Fujimoto, T.; Kuwahara, T.; Eguchi, T.; Sakurai, M.; Komori, T.; Iwatsubo, T. Parkinson’s Disease-Associated Mutant LRRK2 Phosphorylates Rab7L1 and Modifies Trans-Golgi Morphology. Biochem. Biophys. Res. Commun. 2018, 495, 1708–1715.
- Wang, S.; Ma, Z.; Xu, X.; Wang, Z.; Sun, L.; Zhou, Y.; Lin, X.; Hong, W.; Wang, T. A Role of Rab29 in the Integrity of the Trans-Golgi Network and Retrograde Trafficking of Mannose-6-Phosphate Receptor. PLoS ONE 2014, 9, e96242.
- Purlyte, E.; Dhekne, H.S.; Sarhan, A.R.; Gomez, R.; Lis, P.; Wightman, M.; Martinez, T.N.; Tonelli, F.; Pfeffer, S.R.; Alessi, D.R. Rab29 Activation of the Parkinson’s Disease-associated LRRK2 Kinase. EMBO J. 2018, 37, 1–18.
- Matsui, T.; Itoh, T.; Fukuda, M. Small GTPase Rab12 Regulates Constitutive Degradation of Transferrin Receptor. Traffic 2011, 12, 1432–1443.
- Rydell, G.E.; Renard, H.; Garcia-Castillo, M.; Dingli, F.; Loew, D.; Lamaze, C.; Römer, W.; Johannes, L. Rab12 Localizes to Shiga Toxin-Induced Plasma Membrane Invaginations and Controls Toxin Transport. Traffic 2014, 15, 772–787.
- Wang, J.; Lau, P.K.; Li, C.W.; Guo, Y. The Clathrin Adaptor Complex-1 and Rab12 Regulate Post-Golgi Trafficking of WT Epidermal Growth Factor Receptor (EGFR). J. Biol. Chem. 2023, 299, 102979.
- Kluss, J.H.; Mazza, M.C.; Li, Y.; Manzoni, C.; Lewis, P.A.; Cookson, M.R.; Mamais, A. Preclinical Modeling of Chronic Inhibition of the Parkinson’s Disease Associated Kinase LRRK2 Reveals Altered Function of the Endolysosomal System in Vivo. Mol. Neurodegener. 2021, 16, 17.
- Ito, K.; Araki, M.; Katai, Y.; Nishimura, Y.; Imotani, S.; Inoue, H.; Ito, G.; Tomita, T. Pathogenic LRRK2 Compromises the Subcellular Distribution of Lysosomes in a Rab12-RILPL1-dependent Manner. FASEB J. 2023, 37, e22930.
- Ito, G.; Tomita, T.; Utsunomiya-Tate, N. LRRK2-Mediated Phosphorylation and Thermal Stability of Rab12 Are Regulated by Bound Nucleotides. Biochem. Biophys. Res. Commun. 2023, 667, 43–49.
- Klinkert, K.; Echard, A. Rab35 GTPase: A Central Regulator of Phosphoinositides and F-actin in Endocytic Recycling and Beyond. Traffic 2016, 17, 1063–1077.
- Bonet-Ponce, L.; Beilina, A.; Williamson, C.D.; Lindberg, E.; Kluss, J.H.; Saez-Atienzar, S.; Landeck, N.; Kumaran, R.; Mamais, A.; Bleck, C.K.E.; et al. LRRK2 Mediates Tubulation and Vesicle Sorting from Lysosomes. Sci. Adv. 2020, 6, eabb2454.
- Bae, E.-J.; Kim, D.-K.; Kim, C.; Mante, M.; Adame, A.; Rockenstein, E.; Ulusoy, A.; Klinkenberg, M.; Jeong, G.R.; Bae, J.R.; et al. LRRK2 Kinase Regulates α-Synuclein Propagation via RAB35 Phosphorylation. Nat. Commun. 2018, 9, 3465.
- Chen, P.-I.; Kong, C.; Su, X.; Stahl, P.D. Rab5 Isoforms Differentially Regulate the Trafficking and Degradation of Epidermal Growth Factor Receptors*. J. Biol. Chem. 2009, 284, 30328–30338.
- Chen, P.-I.; Schauer, K.; Kong, C.; Harding, A.R.; Goud, B.; Stahl, P.D. Rab5 Isoforms Orchestrate a “Division of Labor” in the Endocytic Network; Rab5C Modulates Rac-Mediated Cell Motility. PLoS ONE 2014, 9, e90384.
- Sung, J.Y.; Kim, J.; Paik, S.R.; Park, J.H.; Ahn, Y.S.; Chung, K.C. Induction of Neuronal Cell Death by Rab5A-Dependent Endocytosis of α-Synuclein*. J. Biol. Chem. 2001, 276, 27441–27448.
- Maekawa, T.; Sasaoka, T.; Azuma, S.; Ichikawa, T.; Melrose, H.L.; Farrer, M.J.; Obata, F. Leucine-Rich Repeat Kinase 2 (LRRK2) Regulates α-Synuclein Clearance in Microglia. BMC Neurosci. 2016, 17, 77.
- Heo, H.Y.; Kim, K.-S.; Seol, W. Coordinate Regulation of Neurite Outgrowth by LRRK2 and Its Interactor, Rab5. Exp. Neurobiol. 2010, 19, 97–105.
- Yun, H.J.; Kim, H.; Ga, I.; Oh, H.; Ho, D.H.; Kim, J.; Seo, H.; Son, I.; Seol, W. An Early Endosome Regulator, Rab5b, Is an LRRK2 Kinase Substrate. J. Biochem. 2015, 157, 485–495.
- Mignogna, M.L.; D’Adamo, P. Critical Importance of RAB Proteins for Synaptic Function. Small GTPase 2018, 9, 145–157.
- Lang, J. Molecular Mechanisms and Regulation of Insulin Exocytosis as a Paradigm of Endocrine Secretion. Eur. J. Biochem. 1999, 259, 3–17.
- Geppert, M.; Bolshakov, V.Y.; Siegelbaum, S.A.; Takei, K.; Camilli, P.D.; Hammer, R.E.; Südhof, T.C. The Role of Rab3A in Neurotransmitter Release. Nature 1994, 369, 493–497.
- Geppert, M.; Goda, Y.; Stevens, C.F.; Südhof, T.C. The Small GTP-Binding Protein Rab3A Regulates a Late Step in Synaptic Vesicle Fusion. Nature 1997, 387, 810–814.
- Riedel, D.; Antonin, W.; Fernandez-Chacon, R.; de Toledo, G.A.; Jo, T.; Geppert, M.; Valentijn, J.A.; Valentijn, K.; Jamieson, J.D.; Südhof, T.C.; et al. Rab3D Is Not Required for Exocrine Exocytosis but for Maintenance of Normally Sized Secretory Granules. Mol. Cell Biol. 2002, 22, 6487–6497.
- Schlüter, O.M.; Schmitz, F.; Jahn, R.; Rosenmund, C.; Südhof, T.C. A Complete Genetic Analysis of Neuronal Rab3 Function. J. Neurosci. 2004, 24, 6629–6637.
- Persoon, C.M.; Hoogstraaten, R.I.; Nassal, J.P.; van Weering, J.R.T.; Kaeser, P.S.; Toonen, R.F.; Verhage, M. The RAB3-RIM Pathway Is Essential for the Release of Neuromodulators. Neuron 2019, 104, 1065–1080.e12.
- Encarnação, M.; Espada, L.; Escrevente, C.; Mateus, D.; Ramalho, J.; Michelet, X.; Santarino, I.; Hsu, V.W.; Brenner, M.B.; Barral, D.C.; et al. A Rab3a-Dependent Complex Essential for Lysosome Positioning and Plasma Membrane Repair. J. Cell Biol. 2016, 213, 631–640.
- Petridi, S.; Middleton, A.C.; Ugbode, C.; Fellgett, A.; Covill, L.; Elliott, C. In Vivo Visual Screen for Dopaminergic Rab ↔ LRRK2-G2019S Interactions in Drosophila Discriminates Rab10 from Rab3. G3 Genes Genomes Genet. 2020, 10, 1903–1914.
- Nirujogi, R.S.; Tonelli, F.; Taylor, M.; Lis, P.; Zimprich, A.; Sammler, E.; Alessi, D.R. Development of a Multiplexed Targeted Mass Spectrometry Assay for LRRK2-Phosphorylated Rabs and Ser910/Ser935 Biomarker Sites. Biochem. J. 2021, 478, 299–326.
- Hatoyama, Y.; Homma, Y.; Hiragi, S.; Fukuda, M. Establishment and Analysis of Conditional Rab1 and Rab5 Knockout Cells by Using the Auxin-Inducible Degron System. J. Cell Sci. 2021, 134, jcs259184.
- Tomás, M.; Martínez-Alonso, E.; Martínez-Martínez, N.; Cara-Esteban, M.; Martínez-Menárguez, J.A. Fragmentation of the Golgi Complex of Dopaminergic Neurons in Human Substantia Nigra: New Cytopathological Findings in Parkinson’s Disease. Histol. Histopathol. 2020, 36, 47–60.
- Fan, Y.; Nirujogi, R.S.; Garrido, A.; Ruiz-Martínez, J.; Bergareche-Yarza, A.; Mondragón-Rezola, E.; Vinagre-Aragón, A.; Croitoru, I.; Pagola, A.G.; Markinez, L.P.; et al. R1441G but Not G2019S Mutation Enhances LRRK2 Mediated Rab10 Phosphorylation in Human Peripheral Blood Neutrophils. Acta Neuropathol. 2021, 142, 475–494.
- Wang, X.; Negrou, E.; Maloney, M.T.; Bondar, V.V.; Andrews, S.V.; Montalban, M.; Llapashtica, C.; Maciuca, R.; Nguyen, H.; Solanoy, H.; et al. Understanding LRRK2 Kinase Activity in Preclinical Models and Human Subjects through Quantitative Analysis of LRRK2 and PT73 Rab10. Sci. Rep. 2021, 11, 12900.
- Petropoulou-Vathi, L.; Simitsi, A.; Valkimadi, P.-E.; Kedariti, M.; Dimitrakopoulos, L.; Koros, C.; Papadimitriou, D.; Papadimitriou, A.; Stefanis, L.; Alcalay, R.N.; et al. Distinct Profiles of LRRK2 Activation and Rab GTPase Phosphorylation in Clinical Samples from Different PD Cohorts. npj Park. Dis. 2022, 8, 73.
- Khan, S.S.; Sobu, Y.; Dhekne, H.S.; Tonelli, F.; Berndsen, K.; Alessi, D.R.; Pfeffer, S.R. Pathogenic LRRK2 Control of Primary Cilia and Hedgehog Signaling in Neurons and Astrocytes of Mouse Brain. eLife 2021, 10, e67900.
- Waschbüsch, D.; Purlyte, E.; Pal, P.; McGrath, E.; Alessi, D.R.; Khan, A.R. Structural Basis for Rab8a Recruitment of RILPL2 via LRRK2 Phosphorylation of Switch 2. Structure 2020, 28, 406–417.e6.
- Boecker, C.A.; Goldsmith, J.; Dou, D.; Cajka, G.G.; Holzbaur, E.L.F. Increased LRRK2 Kinase Activity Alters Neuronal Autophagy by Disrupting the Axonal Transport of Autophagosomes. Curr. Biol. 2021, 31, 2140–2154.e6.
- Kingwell, K. LRRK2-Targeted Parkinson Disease Drug Advances into Phase III. Nat. Rev. Drug Discov. 2023, 22, 3–5.
- Jennings, D.; Huntwork-Rodriguez, S.; Vissers, M.F.J.M.; Daryani, V.M.; Diaz, D.; Goo, M.S.; Chen, J.J.; Maciuca, R.; Fraser, K.; Mabrouk, O.S.; et al. LRRK2 Inhibition by BIIB122 in Healthy Participants and Patients with Parkinson’s Disease. Mov. Disord. 2023, 38, 386–398.
- Jennings, D.; Huntwork-Rodriguez, S.; Henry, A.G.; Sasaki, J.C.; Meisner, R.; Diaz, D.; Solanoy, H.; Wang, X.; Negrou, E.; Bondar, V.V.; et al. Preclinical and Clinical Evaluation of the LRRK2 Inhibitor DNL201 for Parkinson’s Disease. Sci. Transl. Med. 2022, 14, eabj2658.
More