Cardiorenal syndrome type 4 (CRS type 4) occurs when chronic kidney disease (CKD) leads to cardiovascular damage, resulting in high morbidity and mortality rates. Mitochondria, vital organelles responsible for essential cellular functions, can become dysfunctional in CKD. This dysfunction can trigger inflammatory responses in distant organs by releasing Damage-associated molecular patterns (DAMPs). These DAMPs are recognized by immune receptors within cells, including Toll-like receptors (TLR) like TLR2, TLR4, and TLR9, the nucleotide-binding domain, leucine-rich-containing family pyrin domain-containing-3 (NLRP3) inflammasome, and the cyclic guanosine monophosphate (cGMP)–adenosine monophosphate (AMP) synthase (cGAS)–stimulator of interferon genes (cGAS-STING) pathway. Activation of these immune receptors leads to the increased expression of cytokines and chemokines. Excessive chemokine stimulation results in the recruitment of inflammatory cells into tissues, causing chronic damage. Experimental studies have demonstrated that chemokines are upregulated in the heart during CKD, contributing to CRS type 4.
- chronic kidney disease
- cardiorenal syndrome type 4
- mitochondria
- innate immune response
- chemokines
- inflammation
1. Cardiorenal Syndrome Overview
The Kidney-Heart Crosstalk
The causes of CRS type 4 are multifactorial and encompass a range of factors, including hemodynamic alterations, dysregulation of neurohormonal responses, overactivation of the renin–angiotensin–aldosterone system (RAAS), anemia, and pressure overload, among others [11,17,27][17][19][20]. Additionally, as renal function declines, the accumulation of uremic toxins becomes a significant concern [28,29][21][22]. These factors have garnered attention for their roles in inducing cardiovascular alterations secondary to CKD. Consequently, uremic patients often experience endothelial dysfunction, a prevalent complication of CKD [30,31][23][24]. Moreover, kidney–heart crosstalk is mediated by the systemic trafficking of extracellular vesicles (EV). This concept gains support from discovering EV-containing proteins in cardiac tissue that are not typically found in the heart but show increased presence in the kidney [32][25]. Such interorgan communication can shed light on heart dysregulation in the context of CKD. In response to proinflammatory insults, proximal tubule cells release exosomes, a specific type of EV, which carry dysregulated micro ribonucleic acids (RNAs) associated with the regulation of proinflammatory and profibrotic pathways, as well as dysregulated mitochondrial RNAs [33][26]. As CKD progresses, uremic toxins continue to accumulate, exacerbating inflammation and oxidative stress in the kidney [28,34][21][27]. This accumulation likely induces inflammasome activation and pyroptosis by releasing cellular and mitochondrial components [35][28]. Notably, mitochondrial components are identified as mitochondrial damage-associated molecular patterns (mtDAMPs), and they trigger inflammatory and immune responses, contributing to inflammation in various organs [36,37,38][29][30][31]. Mitochondrial DAMPs stimulate innate immune signaling responses in different cardiac cell lineages, triggering the activation of transcription factors such as the nuclear factor-kappa B (NF-κB), a crucial factor for inflammation [39,40][32][33]. In addition, mitochondria provide an assembly platform for signaling innate immune responses, contributing to additional inflammatory responses [41][34]. The main innate immune responses that are activated during the cardiorenal association include Toll-like receptors (TLRs), the nucleotide-binding domain-like receptor family pyrin domain-containing protein 3 (NLRP3) inflammasome, and cyclic guanosine monophosphate (cGMP)–adenosine monophosphate (AMP) synthase (cGAS)–stimulator of interferon genes (STING) [42,43,44,45,46][35][36][37][38][39]. TLRs, NLRP3, and cGAS-STING pathways produce the upregulation of cytokines, vasoactive substances, chemokines, and inflammatory responses [46,47,48][39][40][41]. Chemokine’s overstimulation produces the recruitment of leukocytes to tissues and dysregulated infiltration, leading to chronic cardiac damage [49,50,51][42][43][44]. Interestingly, experimental studies have shown that chemokines are upregulated in the heart during CKD, establishing a link for CRS type 4 development [52,53][45][46]. Chemokine inhibitors have shown promise in reducing chronic inflammation and preventing cardiac and cardiorenal impairment [52,53,54,55][45][46][47][48]. Despite these advancements, the molecular mechanisms underlying how mtDAMPs are released by the kidneys in CKD may trigger innate immune pathways in the heart, ultimately leading to chemokine overactivation and the development of CRS type 4, remain poorly understood.2. Mitochondrial Dysfunction and Inflammatory Alterations in CRS Type 4
Mitochondria are versatile organelles with diverse roles encompassing biosynthesis, metabolism, calcium (Ca2+) regulation, inflammation, and cell death, among other crucial cellular processes [56][49]. Importantly, mitochondria are also the primary source of reactive oxygen species (ROS), particularly in complexes I and III of the electron transport system (ETS) [57][50]. At moderate levels, these ROS function as secondary messengers, governing intracellular signal transduction cascades (Figure 1A) [58,59,60][51][52][53]. However, excess ROS production, often associated with reduced oxidative phosphorylation and ETS activity, leads to oxidative stress (Figure 1B). Notably, due to their high energy demands, the heart and kidneys have a dense population of mitochondria [58,61,62,63][51][54][55][56]. Consequently, mitochondrial dysfunction serves as a potent trigger for the development of renal, cardiac, and cardiorenal diseases.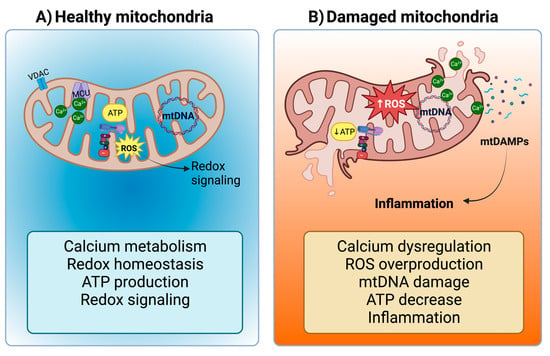
2.1. Mitochondrial Dysfunction in CKD Activates the NLRP3-NF-κB Pathway
2.2. The Release of mtDAMPs during CKD and the Establishment of CRS Type 4
In the context of CKD, inflammation follows a distinct trajectory. Initially, it is characterized by functional alterations, including glomerular hyperfiltration, which serves as a compensatory mechanism [113,114][97][98]. However, structural changes emerge as time progresses, giving rise to a cascade of complications. These structural transformations encompass proteinuria, interstitial nephritis, tubular epithelial–mesenchymal transition, nephron fibrosis, and scarring [65,115,116][58][99][100]. The structural changes are attributed to elevated circulating levels of various molecules, including fibrinogen, TNF-α, and IL-1β and IL-6. These molecules trigger an inflammatory response and promote the secretion of fibrotic mediators [117,118][101][102]. Consequently, inflammation leads to a reduction in mitochondrial ATP production, mitochondrial uncoupling, and an increase in ROS, culminating in oxidative stress and mitochondrial damage [119,120][103][104]. In response to mitochondrial uncoupling or oxidative stress, cells activate apoptosis, a programmed type of cell death. Apoptosis entails the release of mitochondrial proteins to the cytosol. This process is initiated by the oligomerization of effector Bcl2-family proteins such as B-cell lymphoma 2 (BCL-2)-associated X (BAX) and BCL-2 antagonist/killer 1 (BAK). These proteins form oligomers that promote the permeabilization of the mitochondrial outer membrane (MOM). As a result, mtDAMPs are released from both the intermembrane mitochondrial space and the matrix to the cytosol [121,122][105][106]. Another main form of cell death is necrosis, characterized by the plasma membrane’s rupture, which releases intracellular contents. The necrotic process is regulated, and it encompasses various types of necrosis, including ferroptosis, necroptosis, and pyroptosis. Each of these distinct mechanisms contributes significantly to various kidney diseases, either by directly affecting kidney cells or by recruiting immune cells and triggering inflammatory responses [123][107].2.3. NLRP3-NF-κB Pathway Activation in the Heart by CKD-Derived mtDAMPs and ROS
One of the different important stimuli for ROS production and immune activation in CKD occurs following the dissociation of the thioredoxin (TRX) complex from the thioredoxin-interacting protein (TXNIP), ultimately leading to the activation of the NLRP3 inflammasome [135,136][108][109]. TRX are ubiquitously present redox-active proteins known for their antioxidant and anti-inflammatory properties. Elevated ROS levels disrupt the TRX complex, causing TXNIP to bind to the leucine-rich repeat region of NLRP3, consequently activating the inflammasome [137][110]. In the context of CVD, several CKD-related alterations, including increased levels of angiotensin II levels, ROS production, reduced activity of antioxidant enzymes, and inflammation, contribute to decreased levels of thioredoxins [138,139,140][111][112][113]. Furthermore, the depletion of mitochondrial TRX2 in cardiomyocytes leads to hypertrophy and disrupts mitochondrial respiratory function by reducing AMPK activity [141][114]. Impaired mitochondrial function and the activation of the NLRP3 inflammasome are associated with decreased levels of TRX2 during myocardial ischemia–reperfusion injury [142][115]. Intriguingly, elevated levels of NLRP3 and IL-1β observed in patients with coronary artery disease exhibit an inverse association with the expression and protein levels of TXNIP and TRX1 [143][116]. The rise in ROS levels due to disturbances in redox balance can also lead to the oxidation of mtDNA, a mtDAMP that activates the NLRP3 inflammasome [146][117]. Additionally, ROS can directly activate NLRP3 inflammasome in CVD [147][118]. These ROS may especially damage cardiomyocytes by activating the NF-κB pathway and the NLRP3 inflammasome [148][119]. In addition, activation of the NLRP3-NF-κB-ROS pathway in CKD not only initiates inflammation but also triggers additional mechanisms contributing to cardiorenal disturbances. For instance, ROS levels may activate the transforming growth factor beta (TGFβ-1), a key player in cardiac fibrosis [149][120]. TGFβ-1, in turn, further elevates ROS levels, promoting the activation of intracellular Smad pathways, leading to fibrosis, and decreasing antioxidant enzyme levels [149,150][120][121]. The consequence of reduced levels of antioxidant enzyme levels following kidney injury is the promotion of oxidative stress. On the other hand, TNF-α, a pro-inflammatory cytokine known to induce cardiac hypertrophy, fibrosis, dysfunction under pressure overload, and chronic heart injuries [151[122][123],152], may trigger the activation of NLRP3 through the elevation of ROS levels [153,154][124][125]. This phenomenon can be elucidated by the chronic exposure of cells to TNF-α, which sets off p38-mitogen-activated protein kinases (MAPK) signaling, instigates inflammatory phenotypes, and suppresses the expression of antioxidant genes, resulting in an increase in ROS levels [155][126]. Furthermore, the exposure of fibroblasts and human immune cells to TNF-α, in combination with oxidative stress, may prompt the degradation of the IκBα subunit by the IκB Kinase (IKK). This degradation event, in turn, leads to NF-κB activation and the transcription of genes associated with proinflammatory cytokines, chemokines, and NLRP3 inflammasome [156,157][127][128].2.4. Involvement of NLRP3 Inflammasome and Toll-like Receptors 2 and 4 in CRS Type 4
Overactivation of NLRP3 inflammasome has been linked to myocardial fibrosis, hypertrophy, and cardiac dysfunction [154,161][125][129]. The upregulation of NLRP3, IL-1β, and IL-18 in the heart during CKD is closely associated with exposure to DAMPs, thus highlighting the cardiorenal connection [146,162][117][130]. DAMPs and mtDAMPs can trigger NLRP3 activation through TLRs, including uremic toxins, mitochondrial components released due to defects in membrane integrity, mitochondrial ROS, and cardiolipin [45,131][38][131]. TLRs are type I integral transmembrane proteins composed of three main components: an ectodomain with leucine-reach repeats (LRRs), a transmembrane domain, and a cytoplasmic Toll/IL-1 receptor (TIR) domain. LRRs recognize PAMPs or DAMPs, while the TIR domain initiates the downstream signaling [128,163][132][133]. The primary functions of TLRs include stimulating phagocytosis and mediating inflammation by sensing molecules from damaged cells [164][134]. The activation of TLRs by ligands results in the dimerization of TLRs’ cytoplasmic signaling domains [165][135]. This TIR-TIR complex initiates downstream signaling by recruiting specific adaptor molecules [166][136]. DAMPs and mtDAMPs, such as debris from apoptotic and necrotic cells, inflammatory factors, nucleic acid fragments, oxidative products, and uremic toxins generated during renal damage can activate classical TLR2 and TLR4 pathways in the heart [51,71][44][61]. Upon recognition of DAMPs and mtDAMPs by TLR2 and TLR4, they stimulate macrophages to produce inflammatory cytokines [145,167,168][137][138][139]. TLR2 and TLR4 rely on adaptor molecules, with TLR2 engaging myeloid differentiation factor 88 (MyD88) and TLR4 utilizing Toll/IL-1 receptor (TIR) domain-containing adaptor inducing interferon beta (TRIF) [166][136]. Activation of the MyD88-dependent pathway involves the participation of IL-1 receptor-associated kinases (IRAK), including IRAK1 and IRAK4, TNF receptor-associated factor 6 (TRAF-6), and MAPK. These events culminate in the activation of the transcription factor NF-κB, leading to the production of proinflammatory cytokines such as pro-IL-1β and pro-IL-18 [71,169][61][140]. Sustained inflammation in the kidney may lead to the activation of TLR2 and TLR4 in the heart, thereby contributing to CRS type 4. Supporting this notion, the deletion of both TLRs during unilateral kidney ischemia/reperfusion has demonstrated a reduction in cardiac hypertrophy markers such as B-type natriuretic peptide and α-actin. This suggests that sustained inflammation in the kidney can upregulate TLRs in the heart [170][141]. Another set of activators for TLR2 and TLR4 includes HSP proteins. HSPs are intracellular chaperones with a pivotal role in stress responses. In particular, HSP70 has been identified in the extracellular medium, where it is recognized as a DAMP and activates immune cells. This activation results in the secretion of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α [172][142]. The release mechanism of extracellular HSP70 involves a membrane-associated form [173][143]. In the context of CKD, HSP70 levels appear elevated in the urine and serum of patients, which is closely associated with inflammation and immune responses [174,175][144][145]. Therefore, maintaining regular levels of HSP90 is necessary to prevent cardiovascular disturbances. However, it is important to note that an excessive increase in this protein could also potentially favor the development of CRS type 4. In summary, TLR2 and TLR4 activation and downstream inflammatory signaling are central factors contributing to cardiac disorders during CKD, ultimately promoting the establishment of CRS type 4 (Figure 32).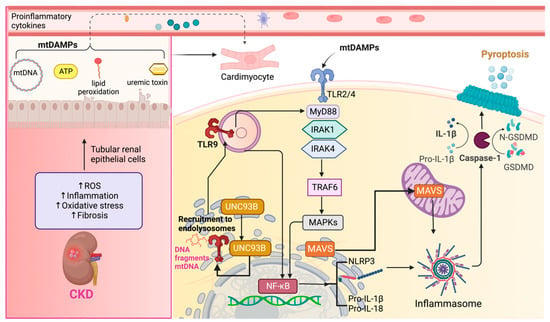
2.5. Role of TLR9 in Inflammation and Its Implication in CRS Type 4
Another TLR known to induce inflammation through the adaptor molecule MyD88 is TLR9 [166][136]. TLR9, primarily localized in endolysosomes, is associated with activating p38 MAPK signaling [179][146]. The inflammatory response initiated by TLR9 is triggered by DNA fragments rich in unmethylated cysteine–phosphate–guanine motifs, with mtDNA being a notable example [180][147]. These DNA fragments can be internalized in various tissues by dendritic cells and macrophages [162][130] and subsequently delivered to endolysosomes, where they activate TLR9 [166][136]. Upon activation, TLR9 interacts with the endoplasmic reticulum (ER) membrane protein UNC93B, facilitating its transportation to the endolysosomal compartment [181,182][148][149] (Figure 3). TLR9 activation leads to two distinct pathways [183][150]: one is associated with the transcriptional activation of proinflammatory cytokines, requiring the involvement of NF-κB, while the other relates to the activation of type I interferon genes [184][151]. Activation of TLR9 increases in renal proximal tubular cells following ischemic injury, initiating a cascade of events that promote inflammation, apoptosis, and necrosis through NF-κB and caspase-dependent pathways [186][152]. Because of apoptosis or necrosis, renal DAMPs are released. These DAMPs may be exposed to the cell surface and released into the extracellular space, acting as potent inflammation triggers [125][153]. Renal DAMPs have the potential to activate TLR9 in cardiac cells, inducing oxidative stress and inflammatory responses, which can lead to the release of mtDNA or other mtDAMPs within cardiomyocytes. This process has been observed in mice cardiomyocytes, where the release of mtDNA after myocardial injury activated NF-κB through TLR9, ultimately contributing to cell death [187][154].2.6. Extracellular Vesicles (EV) and Their Role in Inflammation
Exogenous mtDNA and other signaling molecules may also be transported into other cells, including cardiac cells, through extracellular vesicles (EVs). These EVs, such as exosomes, microvesicles, and apoptotic bodies, carry various cargo, including nucleic acids, proteins, and metabolic intermediaries [190,191][155][156]. Apoptotic bodies, in particular, contain fragments of nucleic acids, lipids, proteins, and organelles [192][157]. Notably, apoptotic cells release ATP, which can serve as a signaling molecule by binding to purinergic receptors on cell membranes, activating intracellular signaling pathways, and potentially inflammasome assembly.2.7. The Role of Autophagy and Mitophagy in NLRP3 Signaling Pathway in CRS Type 4
Autophagy consists of vesicular sequestration of cellular components, inducing their degradation and further recycling [196][158]. This process comprises initiation, elongation, fusion, and degradation and is regulated by the phosphoinositide-3 kinase (PI3K) and Unc-51-like kinase (ULK) complexes [197][159]. The activation of ULK depends on the AMPK protein that phosphorylates and inhibits the mammalian target of Rapamycin complex 1 (mTORC1). PI3K is activated after the autophagic protein Beclin is disassembled from Bcl2, forming the PI3K complex to produce phosphatidyl inositol triphosphate (PI3P). On the other hand, mitophagy is a specialized form of autophagy that removes damaged mitochondria and is crucial for immune system vigilance and mitochondrial quality control. Mitophagy occurs when the mitochondrial membrane potential (ΔΨ) is disrupted and involves PTEN-induced kinase (PINK) and E3 ubiquitin ligase (Parkin) proteins. Both in autophagy and mitophagy processes, sequestosome p62 proteins (p62) are necessary for degradation because these proteins recognize and ubiquitinate damaged organelle and protein aggregates. Moreover, the microtubule-associated protein 1A/1B-light chain 3 (LC3) is involved in the elongation of autophagosomes [198][160]. Mitophagy’s role in NLRP3 inflammasome activation is shown by removing autophagy-related proteins, causing the accumulation of damaged mitochondria, and increasing mtDAMPs production [199][161]. For example, it has been demonstrated that NF-KB restricts NLRP3 inflammasome activation through p62-dependent mitophagy; conversely, the absence of p62 promotes greater mitochondrial damage and increased inflammation [200][162].2.8. MAVS and NLRP3-NF-kB Signaling in CRS Type 4
Another role of mitochondria in NLRP3 activation is associated with mitochondrial antiviral proteins (MAVS). MAVS comprises an N-terminal CARD-like domain and a C-terminal transmembrane domain, essential for MAVS signaling. Notably, the transmembrane domain targets MAVS to the mitochondria in the MOM [207][163]. The latter allows MAVS to participate in the relocalization and association of NLRP3 with ER and mitochondria organelle clusters [137][110]. This facilitates NLRP3 oligomerization [208,209][164][165]. Low active caspase-1, IL-1β, and IL-18 levels induce cytokine production, but higher levels of these molecules can induce cell death by apoptosis or pyroptosis. When NLRP3 is activated and associated with MAVS, it leads to pyroptosis [211][166]. Pyroptosis is a caspase-1-dependent death mediated by the cleavage of gasdermin D by caspase-1 and the subsequent formation of stable pores in the cell membrane [212,213][167][168]. The pores formed by gasdermin D proteins promote cell swelling and lytic cell death, releasing cytosolic contents into the extracellular space that act as DAMPs [214][169]. Also, pyroptosis is regulated through the NLRP3 inflammasome [215][170]. It could be associated with the release of DAMPs from renal cells, which may activate inflammatory processes in other organs, such as the heart.3. The Role of the cGAS-STING Pathway in CRS Type 4
3.1. The cGAS-STING Pathway
The cGAS-STING pathway plays a pivotal role in mediating inflammation in response to infections, cellular stress, and tissue damage [218][171]. cGAS activity is triggered by interactions with various ligands, including double-strand DNA (dsDNA), neutrophil DNA–protein complexes, and mtDNA in mammals [218,219][171][172]. When cGAS interacts with these ligands, it generates a product known as 2′3′cyclic GMP-AMP [220][173]. This cyclic GMP-AMP molecule then binds to the STING protein located in the ER membrane [221][174]. The downstream signaling cascade begins with the translocation of STING from the ER to the Golgi apparatus, facilitated by the ER-to-Golgi transport machinery, specifically the ER–Golgi intermediate compartment (ERGIC) [218,221][171][174]. This translocation of STING is a critical step in activating the immune signaling pathway [222,223][175][176]. Once in perinuclear compartments, STING forms a complex with TRAF family member-associated NF-κB activator (TANK)-binding kinase (TBK1) [224][177]. TBK1, in turn, phosphorylates transcription factors, including the interferon regulatory factor 3 (IRF3) and NF-κB [218][171].3.2. The Activation of the cGAS-STING-NF-κB Axis by mtDNA Release in CKD
CKD has been strongly linked to the activation of the cGAS-STING pathway. For instance, in a study conducted by Chung et al. [226][178], a positive correlation was observed between CKD-induced fibrosis and the expression of cGAS and STING in over 400 kidney tissue samples. Experimental models of diabetic kidney disease and Alport syndrome have shown that the cGAS-STING pathway plays a significant role in the development and progression of glomerular damage by regulating inflammation [227][179]. Specifically, this pathway is associated with cell damage and chronic inflammation, resulting in the production of inflammatory cytokines and interferons [228][180]. In CKD, an oxidative stress state is closely related to renal functional and structural alterations, primarily through mitochondrial dysfunction and increased production of ROS [8]. Notably, the plasma of patients receiving platinum-based nephrotoxic anticancer therapy showed elevated levels of mtDNA in plasma, suggesting that STING signaling might be activated through this mechanism [229][181]. The generation of ROS and Ca2+ ion accumulation can trigger the opening of the mitochondrial permeability transition pore (mPTP), resulting in the loss of ΔΨ, uncoupling of the ETS, and the release of proapoptotic factors like cytochrome c, which can lead to apoptosis or necrosis. During apoptosis, macropores form in the MOM due to the regulation of BAX and BAK [231,232,233][182][183][184]. These BAX-mediated pores in the MOM allow the inner membrane to herniate, leaking mtDNA and other mitochondrial matrix components in the cytoplasm. In the context of cisplatin-induced nephrotoxicity, it has been suggested that mitochondrial permeabilization induced by BAX pores in the MOM can activate the cGAS-STING pathway, thus triggering inflammation [225][185]. Small-molecule STING inhibitors, such as H151, have shown promise in ameliorating renal function, kidney morphology, inflammation, and mitochondrial alterations following cisplatin-induced nephrotoxicity [229][181]. Additionally, activation of the cGAS-STING pathway has been observed in diabetic kidney disease resulting from mitochondrial damage [235][186].3.3. The Activation of the cGAS-STING-NF-κB Axis by mtDNA Release in CRS Type 4
Activation of the immune response in CRS type 4 has been linked to the escape of mtDNA into the cytoplasm, thereby triggering the cGAS-STING pathway [226][178]. In experimental diabetic cardiomyopathy, the release of mtDNA into the cytosol of heart cells induces inflammation through the cGAS-STING pathway, activating downstream genes, including IRF3, NF-κB, IL-18, and IL-1β [219][172]. IL-1β, in particular, can potentially disrupt mitochondrial homeostasis by amplifying immune reactions through its activation of cGAS via mtDNA [244,245][187][188]. In experimental models of uremic cardiomyopathy, mitochondrial oxidative stress emerges as a consequence of CKD. Oxidative stress triggers the voltage-dependent anion channel (VDAC)-mediated MOM permeabilization, leading to the release of mtDNA and subsequently activating the STING-NF-κB pathway within the heart [41][34]. DNA fragments released from metabolic organs, originating from the body’s own cells, promote chronic inflammation as they serve as endogenous ligands for the cGAS-STING pathway [227][179].4. Chemokines Activation and the Pathophysiology of CRS Type 4
4.1. Chemokines Overview
4.2. The Role of Chemokines and Receptors in the Pathophysiology of CKD
In a healthy kidney, various cell types, including endothelial cells, podocytes, mesangial cells, tubular epithelial cells, and interstitial fibroblasts, typically produce low levels of inflammatory chemokines [260,261][197][198]. In patients with CKD, these chemokines are predominately induced by pro-inflammatory cytokines and ROS [262][199]. The primary role of chemokines in the kidney is to facilitate the recruitment of leukocytes and T cells, which play a central in interstitial fibrosis and the progression of CKD [260,263][197][200]. Other factors contributing to chemokine activation in CKD include uremic toxins, cyclic adenosine monophosphate (cAMP), growth factors, lipopolysaccharides, low-density lipoprotein (LDL), IFN-γ, and vasoactive substances [253,262,264][199][201][202]. These factors can further upregulate chemokines by influencing NF-κB and other transcription factors [150][121]. Consequently, an excess of damaging stimuli in CKD can lead to the overstimulation of chemokines, accelerating disease progression.4.2.1. Monocyte Chemoattractant Protein-1 (MCP-1)/CCL2 and CCR2 Receptor in CKD
CCL-2 is a well-studied chemokine in cardiac and renal diseases, known for its ability to attract monocytes, T lymphocytes, and natural killer cells [250][191]. Excessive activation of CCL2 leads to an overwhelming cellular infiltration and prolonged inflammatory response, exacerbating tissue damage and affecting kidney function [265,266][203][204]. Upregulation of CCL2 by NF-κB has been linked to tubulointerstitial injury in proteinuric renal disease [267][205]. Conversely, reducing protein accumulation in renal disease has been shown to decrease CCL2 levels [268][206]. In advanced CKD, the TGF-β/Smad2,3 pathway activation induces CCL2 expression in renal cells, resulting in a chemotactic effect on macrophages [269][207]. Likewise, in the UUO model, a well-established model for studying fibrosis in CKD, a wide expression of CCL2 is observed, leading to macrophage infiltration, tubulointerstitial CCL2 expression, leading to macrophage infiltration via a TGF-β/Smad3-dependent signaling pathway [270,271][208][209]. Therefore, CCL2 plays a pivotal role in progressive interstitial fibrosis in CKD.4.2.2. C-C Motif Chemokine 8 (CCL8/MCP-2) in CKD
CCL8 is a CC chemokine that plays a pivotal role in attracting inflammatory monocytes and T lymphocytes in various pathological conditions [278,279][210][211]. In advanced CKD and fibrosis-related human glomerulopathies [280][212], CCL8 levels significantly increase, primarily due to the activation of the TGF-β pathway. Consequently, inhibiting CCL8 has been proposed as a preventive therapy against fibrosis in CKD. In the mice-UUO model, functional blockade of CCL8 with a monoclonal antibody has been shown to prevent fibrosis and apoptosis in renal cells [50][43].4.2.3. Chemokine Interferon-γ-Inducible Protein 10 (IP-10)/Chemokine (C-X-C Motif) Ligand (CXCL)10 in CKD
CXCL10, a member of the CXC chemokine family, exerts its biological functions by binding to the CXCR3 receptor [283][213]. CXCR3 is expressed in T lymphocytes, natural killer (NK) cells, inflammatory dendritic cells, macrophages, and B cells [284][214]. CXCL10 is involved in chemotaxis, apoptosis induction, cell growth regulation, and angiostatic effects. It is primarily secreted by leukocytes, activated neutrophils, eosinophils, epithelial cells, and endothelial cells in response to IFN-γ [273][215]. Once activated, CXCL10 attracts Th1 lymphocytes, monocytes, T cells, and NK cells [273,283][213][215]. Interstitial CXCR3 has been implicated in the progressive loss of renal function in human glomerular diseases [260][197]. As a consequence of CKD, inflammatory processes can also manifest in the heart, resulting in significant alterations, including heart failure, coronary artery disease, arrhythmias, and sudden cardiac death. This can ultimately lead to the development of CRS type 4.5. Conclusions
CKD induces hemodynamic and metabolic changes that lead to mitochondrial damage, causing the release of various components into the peripheral circulation. These mitochondrial components activate inflammatory signaling pathways in organs like the heart, resulting in the upregulation of inflammatory genes, including chemokines and cytokines, further exacerbating damage. Chemokines play a pivotal role in attracting inflammatory cells, thereby intensifying inflammation, and contributing to the development of CRS type 4 development.References
- Bright, R. Cases and Observations, Illustrative of Renal Disease, Accompanied with the Secretion of Albuminous Urine. Guy’s Hosp. Rep. 1836, 1, 338–379.
- Ronco, C.; McCullough, P.; Anker, S.D.; Anand, I.; Aspromonte, N.; Bagshaw, S.M.; Bellomo, R.; Berl, T.; Bobek, I.; Cruz, D.N.; et al. Cardio-Renal Syndromes: Report from the Consensus Conference of the Acute Dialysis Quality Initiative. Eur. Heart J. 2010, 31, 703–711.
- Davenport, A.; Anker, S.D.; Mebazaa, A.; Palazzuoli, A.; Vescovo, G.; Bellomo, R.; Ponikowski, P.; Anand, I.; Aspromonte, N.; Bagshaw, S.; et al. ADQI 7: The Clinical Management of the Cardio-Renal Syndromes: Work Group Statements from the 7th ADQI Consensus Conference. Nephrol. Dial. Transplant. 2010, 25, 2077–2089.
- Rangaswami, J.; Bhalla, V.; Blair, J.E.A.; Chang, T.I.; Costa, S.; Lentine, K.L.; Lerma, E.V.; Mezue, K.; Molitch, M.; Mullens, W.; et al. Cardiorenal Syndrome: Classification, Pathophysiology, Diagnosis, and Treatment Strategies: A Scientific Statement from the American Heart Association. Circulation 2019, 139, E840–E878.
- Yogasundaram, H.; Chappell, M.C.; Braam, B.; Oudit, G.Y. Cardiorenal Syndrome and Heart Failure—Challenges and Opportunities. Can. J. Cardiol. 2019, 35, 1208–1219.
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A Single Number for Advocacy and Communication—Worldwide More than 850 Million Individuals Have Kidney Diseases. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2019, 34, 1803–1805.
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, Regional, and National Burden of Chronic Kidney Disease, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733.
- Fontecha-Barriuso, M.; Lopez-Diaz, A.M.; Guerrero-Mauvecin, J.; Miguel, V.; Ramos, A.M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Tubular Mitochondrial Dysfunction, Oxidative Stress, and Progression of Chronic Kidney Disease. Antioxidants 2022, 11, 1356.
- Andrassy, K.M. Comments on ‘KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease’. Kidney Int. 2013, 84, 622–623.
- Kidney Disease: Improving Global Outcomes (KDIGO) Diabetes Work Group. KDIGO 2020 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int. 2020, 98, S1–S115.
- Tumlin, J.A.; Costanzo, M.R.; Chawla, L.S.; Herzog, C.A.; Kellum, J.A.; McCullough, P.A.; Ronco, C. Cardiorenal Syndrome Type 4: Insights on Clinical Presentation and Pathophysiology from the Eleventh Consensus Conference of the Acute Dialysis Quality Initiative (ADQI). In Contributions to Nephrology; McCullough, P.A., Kellum, J.A., Mehta, R.L., Murray, P.T., Ronco, C., Eds.; S. Karger AG: Basel, Switzerland, 2013; Volume 182, pp. 158–173. ISBN 978-3-318-02406-7.
- Di Lullo, L.; Gorini, A.; Russo, D.; Santoboni, A.; Ronco, C. Left Ventricular Hypertrophy in Chronic Kidney Disease Patients: From Pathophysiology to Treatment. Cardiorenal Med. 2015, 5, 254–266.
- Kumar, S.; Bogle, R.; Banerjee, D. Why Do Young People with Chronic Kidney Disease Die Early? World J. Nephrol. 2014, 3, 143–155.
- Mittalhenkle, A.; Stehman-Breen, C.O.; Shlipak, M.G.; Fried, L.F.; Katz, R.; Young, B.A.; Seliger, S.; Gillen, D.; Newman, A.B.; Psaty, B.M.; et al. Cardiovascular Risk Factors and Incident Acute Renal Failure in Older Adults: The Cardiovascular Health Study. Clin. J. Am. Soc. Nephrol. CJASN 2008, 3, 450–456.
- Hager, M.R.; Narla, A.D.; Tannock, L.R. Dyslipidemia in Patients with Chronic Kidney Disease. Rev. Endocr. Metab. Disord. 2017, 18, 29–40.
- Bansal, N.; Katz, R.; Robinson-Cohen, C.; Odden, M.C.; Dalrymple, L.; Shlipak, M.G.; Sarnak, M.J.; Siscovick, D.S.; Zelnick, L.; Psaty, B.M.; et al. Absolute Rates of Heart Failure, Coronary Heart Disease, and Stroke in Chronic Kidney Disease: An Analysis of 3 Community-Based Cohort Studies. JAMA Cardiol. 2017, 2, 314–318.
- McCullough, P.A. Cardiorenal Syndromes: Pathophysiology to Prevention. Int. J. Nephrol. 2011, 2011, 762590.
- Ling, X.C.; Kuo, K.-L. Oxidative Stress in Chronic Kidney Disease. Ren. Replace. Ther. 2018, 4, 53.
- Da Silva, A.L.P.; da Silva, M.J.V. Type 4 Cardiorenal Syndrome. Rev. Port. Cardiol. 2016, 35, 601–616.
- Rysz, J.; Franczyk, B.; Ławiński, J.; Gluba-Brzózka, A. Oxidative Stress in ESRD Patients on Dialysis and the Risk of Cardiovascular Diseases. Antioxidants 2020, 9, 1079.
- Moradi, H.; Sica, D.A.; Kalantar-Zadeh, K. Cardiovascular Burden Associated with Uremic Toxins in Patients with Chronic Kidney Disease. Am. J. Nephrol. 2013, 38, 136–148.
- Schophuizen, C.M.S.; Wilmer, M.J.; Jansen, J.; Gustavsson, L.; Hilgendorf, C.; Hoenderop, J.G.J.; van den Heuvel, L.P.; Masereeuw, R. Cationic Uremic Toxins Affect Human Renal Proximal Tubule Cell Functioning through Interaction with the Organic Cation Transporter. Pflügers Arch.-Eur. J. Physiol. 2013, 465, 1701–1714.
- Kopel, T.; Kaufman, J.S.; Hamburg, N.; Sampalis, J.S.; Vita, J.A.; Dember, L.M. Endothelium-Dependent and -Independent Vascular Function in Advanced Chronic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2017, 12, 1588–1594.
- Lano, G.; Burtey, S.; Sallée, M. Indoxyl Sulfate, a Uremic Endotheliotoxin. Toxins 2020, 12, 229.
- Claridge, B.; Rai, A.; Fang, H.; Matsumoto, A.; Luo, J.; McMullen, J.R.; Greening, D.W. Proteome Characterisation of Extracellular Vesicles Isolated from Heart. Proteomics 2021, 21, 2100026.
- Ranches, G.; Zeidler, M.; Kessler, R.; Hoelzl, M.; Hess, M.W.; Vosper, J.; Perco, P.; Schramek, H.; Kummer, K.K.; Kress, M.; et al. Exosomal Mitochondrial tRNAs and miRNAs as Potential Predictors of Inflammation in Renal Proximal Tubular Epithelial Cells. Mol. Ther.-Nucleic Acids 2022, 28, 794–813.
- Cozzolino, M.; Mangano, M.; Stucchi, A.; Ciceri, P.; Conte, F.; Galassi, A. Cardiovascular Disease in Dialysis Patients. Nephrol. Dial. Transplant. 2018, 33, iii28–iii34.
- Ding, B.; Ma, G.; Wang, Z.; Liang, W.; Gao, W. Mechanisms of Kidney Cell Pyroptosis in Chronic Kidney Disease and the Effects of Traditional Chinese Medicine. Evid. Based Complement. Altern. Med. 2021, 2021, 1173324.
- Cachofeiro, V.; Goicochea, M.; de Vinuesa, S.G.; Oubiña, P.; Lahera, V.; Luño, J. Oxidative Stress and Inflammation, a Link between Chronic Kidney Disease and Cardiovascular Disease. Kidney Int. 2008, 74, S4–S9.
- Ayoub, K.F.; Pothineni, N.V.K.; Rutland, J.; Ding, Z.; Mehta, J.L. Immunity, Inflammation, and Oxidative Stress in Heart Failure: Emerging Molecular Targets. Cardiovasc. Drugs Ther. 2017, 31, 593–608.
- López-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-García, C.; Valcárcel-Ares, M.N. Mitochondrial Dysfunction and the Inflammatory Response. Mitochondrion 2013, 13, 106–118.
- Schefold, J.C.; Filippatos, G.; Hasenfuss, G.; Anker, S.D.; von Haehling, S. Heart Failure and Kidney Dysfunction: Epidemiology, Mechanisms and Management. Nat. Rev. Nephrol. 2016, 12, 610–623.
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216.
- Sandhir, R.; Halder, A.; Sunkaria, A. Mitochondria as a Centrally Positioned Hub in the Innate Immune Response. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1090–1097.
- Zhao, W.; Zhou, L.; Novák, P.; Shi, X.; Lin, C.B.; Zhu, X.; Yin, K. Metabolic Dysfunction in the Regulation of the NLRP3 Inflammasome Activation: A Potential Target for Diabetic Nephropathy. J. Diabetes Res. 2022, 2022, 2193768.
- Chin, L.-H.; Hsu, Y.-J.; Hsu, S.-C.; Chen, Y.-H.; Chang, Y.-L.; Huang, S.-M.; Tsai, C.-S.; Lin, C.-Y. The Regulation of NLRP3 Inflammasome Expression during the Development of Cardiac Contractile Dysfunction in Chronic Kidney Disease. Oncotarget 2017, 8, 113303–113317.
- Han, W.; Du, C.; Zhu, Y.; Ran, L.; Wang, Y.; Xiong, J.; Wu, Y.; Lan, Q.; Wang, Y.; Wang, L.; et al. Targeting Myocardial Mitochondria-STING-Polyamine Axis Prevents Cardiac Hypertrophy in Chronic Kidney Disease. JACC Basic Transl. Sci. 2022, 7, 820–840.
- Ou, L.; Zhang, A.; Cheng, Y.; Chen, Y. The cGAS-STING Pathway: A Promising Immunotherapy Target. Front. Immunol. 2021, 12, 795048.
- Skopelja-Gardner, S.; An, J.; Elkon, K.B. Role of the cGAS–STING Pathway in Systemic and Organ-Specific Diseases. Nat. Rev. Nephrol. 2022, 18, 558–572.
- Kawai, T.; Akira, S. TLR Signaling. Cell Death Differ. 2006, 13, 816–825.
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 Inflammasome: From a Danger Signal Sensor to a Regulatory Node of Oxidative Stress and Inflammatory Diseases. Redox Biol. 2015, 4, 296–307.
- Hemmers, C.; Schulte, C.; Wollenhaupt, J.; Wong, D.W.L.; Harlacher, E.; Orth-Alampour, S.; Klinkhammer, B.M.; Schirmer, S.H.; Böhm, M.; Marx, N.; et al. Chemokine CCL9 Is Upregulated Early in Chronic Kidney Disease and Counteracts Kidney Inflammation and Fibrosis. Biomedicines 2022, 10, 420.
- Lee, J.; Lee, Y.; Kim, K.-H.; Kim, D.-K.; Joo, K.-W.; Shin, S.-J.; Kim, Y.-S.; Yang, S.-H. Chemokine (C-C Motif) Ligand 8 and Tubulo-Interstitial Injury in Chronic Kidney Disease. Cells 2022, 11, 658.
- Hanna, A.; Frangogiannis, N.G. Inflammatory Cytokines and Chemokines as Therapeutic Targets in Heart Failure. Cardiovasc. Drugs Ther. 2020, 34, 849–863.
- Amador-Martínez, I.; García-Ballhaus, J.; Buelna-Chontal, M.; Cortés-González, C.; Massó, F.; Jaisser, F.; Barrera-Chimal, J. Early Inflammatory Changes and CC Chemokine Ligand-8 Upregulation in the Heart Contribute to Uremic Cardiomyopathy. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21761.
- Mawhin, M.A.; Bright, R.G.; Fourre, J.D.; Vloumidi, E.I.; Tomlinson, J.; Sardini, A.; Pusey, C.D.; Woollard, K.J. Chronic Kidney Disease Mediates Cardiac Dysfunction Associated with Increased Resident Cardiac Macrophages. BMC Nephrol. 2022, 23, 47.
- Noels, H.; Weber, C.; Koenen, R.R. Chemokines as Therapeutic Targets in Cardiovascular Disease: The Road Behind, The Road Ahead. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 583–592.
- Chen, B.; Frangogiannis, N.G. Chemokines in Myocardial Infarction. J. Cardiovasc. Transl. Res. 2021, 14, 35–52.
- Mejia, E.M.; Hatch, G.M. Mitochondrial Phospholipids: Role in Mitochondrial Function. J. Bioenerg. Biomembr. 2016, 48, 99–112.
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Pedraza-Chaverri, J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules 2021, 11, 1144.
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462.
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial Metabolism of Reactive Oxygen Species. Biochemistry 2005, 70, 200–214.
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining Roles of Specific Reactive Oxygen Species (ROS) in Cell Biology and Physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515.
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox Signal. 2016, 25, 119–146.
- Doi, K.; Noiri, E. Mitochondrial Dysfunction in Cardiorenal Syndrome. Antioxid. Redox Signal. 2016, 25, 200–207.
- Shi, S.; Zhang, B.; Li, Y.; Xu, X.; Lv, J.; Jia, Q.; Chai, R.; Xue, W.; Li, Y.; Wang, Y.; et al. Mitochondrial Dysfunction: An Emerging Link in the Pathophysiology of Cardiorenal Syndrome. Front. Cardiovasc. Med. 2022, 9, 837270.
- Brooks, C.; Wei, Q.; Cho, S.-G.; Dong, Z. Regulation of Mitochondrial Dynamics in Acute Kidney Injury in Cell Culture and Rodent Models. J. Clin. Investig. 2009, 119, 1275–1285.
- Aparicio-Trejo, O.E.; Tapia, E.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Mitochondrial Bioenergetics, Redox State, Dynamics and Turnover Alterations in Renal Mass Reduction Models of Chronic Kidney Diseases and Their Possible Implications in the Progression of This Illness. Pharmacol. Res. 2018, 135, 1–11.
- Feehan, K.T.; Gilroy, D.W. Is Resolution the End of Inflammation? Trends Mol. Med. 2019, 25, 198–214.
- Kolaczkowska, E.; Kubes, P. Neutrophil Recruitment and Function in Health and Inflammation. Nat. Rev. Immunol. 2013, 13, 159–175.
- Junho, C.V.C.; Trentin-Sonoda, M.; Panico, K.; dos Santos, R.S.N.; Abrahão, M.V.; Vernier, I.C.S.; Fürstenau, C.R.; Carneiro-Ramos, M.S. Cardiorenal Syndrome: Long Road between Kidney and Heart. Heart Fail. Rev. 2022, 27, 2137–2153.
- Huang, G.; Zhang, Y.; Zhang, Y.; Ma, Y. Chronic Kidney Disease and NLRP3 Inflammasome: Pathogenesis, Development and Targeted Therapeutic Strategies. Biochem. Biophys. Rep. 2023, 33, 101417.
- Zheng, Z.; Xu, K.; Li, C.; Qi, C.; Fang, Y.; Zhu, N.; Bao, J.; Zhao, Z.; Yu, Q.; Wu, H.; et al. NLRP3 Associated with Chronic Kidney Disease Progression after Ischemia/Reperfusion-Induced Acute Kidney Injury. Cell Death Discov. 2021, 7, 324.
- Granata, S.; Zaza, G.; Simone, S.; Villani, G.; Latorre, D.; Pontrelli, P.; Carella, M.; Schena, F.P.; Grandaliano, G.; Pertosa, G. Mitochondrial Dysregulation and Oxidative Stress in Patients with Chronic Kidney Disease. BMC Genom. 2009, 10, 388.
- Jo, E.-K.; Kim, J.K.; Shin, D.-M.; Sasakawa, C. Molecular Mechanisms Regulating NLRP3 Inflammasome Activation. Cell. Mol. Immunol. 2016, 13, 148–159.
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat. Med. 2015, 21, 677–687.
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in Health and Disease. Nature 2012, 481, 278–286.
- Kinoshita, T.; Imamura, R.; Kushiyama, H.; Suda, T. NLRP3 Mediates NF-κB Activation and Cytokine Induction in Microbially Induced and Sterile Inflammation. PLoS ONE 2015, 10, e0119179.
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023.
- Vringer, E.; Tait, S.W.G. Mitochondria and Cell Death-Associated Inflammation. Cell Death Differ. 2023, 30, 304–312.
- Zhong, F.; Liang, S.; Zhong, Z. Emerging Role of Mitochondrial DNA as a Major Driver of Inflammation and Disease Progression. Trends Immunol. 2019, 40, 1120–1133.
- Morgan, M.J.; Liu, Z. Crosstalk of Reactive Oxygen Species and NF-κB Signaling. Cell Res. 2011, 21, 103–115.
- Aparicio-Trejo, O.E.; Tapia, E.; Molina-Jijón, E.; Medina-Campos, O.N.; Macías-Ruvalcaba, N.A.; León-Contreras, J.C.; Hernández-Pando, R.; García-Arroyo, F.E.; Cristóbal, M.; Sánchez-Lozada, L.G.; et al. Curcumin Prevents Mitochondrial Dynamics Disturbances in Early 5/6 Nephrectomy: Relation to Oxidative Stress and Mitochondrial Bioenergetics: Curcumin Prevents Mitochondrial Dynamics Disturbances. BioFactors 2017, 43, 293–310.
- Forbes, J.M.; Thorburn, D.R. Mitochondrial Dysfunction in Diabetic Kidney Disease. Nat. Rev. Nephrol. 2018, 14, 291–312.
- Coughlan, M.T.; Nguyen, T.-V.; Penfold, S.A.; Higgins, G.C.; Thallas-Bonke, V.; Tan, S.M.; Van Bergen, N.J.; Sourris, K.C.; Harcourt, B.E.; Thorburn, D.R.; et al. Mapping Time-Course Mitochondrial Adaptations in the Kidney in Experimental Diabetes. Clin. Sci. 2016, 130, 711–720.
- Hallan, S.; Afkarian, M.; Zelnick, L.R.; Kestenbaum, B.; Sharma, S.; Saito, R.; Darshi, M.; Barding, G.; Raftery, D.; Ju, W.; et al. Metabolomics and Gene Expression Analysis Reveal Down-Regulation of the Citric Acid (TCA) Cycle in Non-Diabetic CKD Patients. EBioMedicine 2017, 26, 68–77.
- Jiménez-Uribe, A.P.; Bellido, B.; Aparicio-Trejo, O.E.; Tapia, E.; Sánchez-Lozada, L.G.; Hernández-Santos, J.A.; Fernández-Valverde, F.; Hernández-Cruz, E.Y.; Orozco-Ibarra, M.; Pedraza-Chaverri, J. Temporal Characterization of Mitochondrial Impairment in the Unilateral Ureteral Obstruction Model in Rats. Free Radic. Biol. Med. 2021, 172, 358–371.
- Aparicio-Trejo, O.E.; Reyes-Fermín, L.M.; Briones-Herrera, A.; Tapia, E.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Protective Effects of N-Acetyl-Cysteine in Mitochondria Bioenergetics, Oxidative Stress, Dynamics and S-Glutathionylation Alterations in Acute Kidney Damage Induced by Folic Acid. Free Radic. Biol. Med. 2019, 130, 379–396.
- Hui, Y.; Lu, M.; Han, Y.; Zhou, H.; Liu, W.; Li, L.; Jin, R. Resveratrol Improves Mitochondrial Function in the Remnant Kidney from 5/6 Nephrectomized Rats. Acta Histochem. 2017, 119, 392–399.
- Correa, F.; Buelna-Chontal, M.; Hernández-Reséndiz, S.; García-Niño, W.R.; Roldán, F.J.; Soto, V.; Silva-Palacios, A.; Amador, A.; Pedraza-Chaverrí, J.; Tapia, E.; et al. Curcumin Maintains Cardiac and Mitochondrial Function in Chronic Kidney Disease. Free Radic. Biol. Med. 2013, 61, 119–129.
- Liu, H.; Li, W.; He, Q.; Xue, J.; Wang, J.; Xiong, C.; Pu, X.; Nie, Z. Mass Spectrometry Imaging of Kidney Tissue Sections of Rat Subjected to Unilateral Ureteral Obstruction. Sci. Rep. 2017, 7, 41954.
- Zhang, W.; Zhou, X.; Yao, Q.; Liu, Y.; Zhang, H.; Dong, Z. HIF-1-Mediated Production of Exosomes during Hypoxia Is Protective in Renal Tubular Cells. Am. J. Physiol. Renal Physiol. 2017, 313, F906–F913.
- Olona, A.; Leishman, S.; Anand, P.K. The NLRP3 Inflammasome: Regulation by Metabolic Signals. Trends Immunol. 2022, 43, 978–989.
- Nishi, H.; Higashihara, T.; Inagi, R. Lipotoxicity in Kidney, Heart, and Skeletal Muscle Dysfunction. Nutrients 2019, 11, 1664.
- Szeto, H.H.; Liu, S.; Soong, Y.; Alam, N.; Prusky, G.T.; Seshan, S.V. Protection of Mitochondria Prevents High-Fat Diet–Induced Glomerulopathy and Proximal Tubular Injury. Kidney Int. 2016, 90, 997–1011.
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.-A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective Fatty Acid Oxidation in Renal Tubular Epithelial Cells Has a Key Role in Kidney Fibrosis Development. Nat. Med. 2015, 21, 37–46.
- Souza, A.C.P.; Bocharov, A.V.; Baranova, I.N.; Vishnyakova, T.G.; Huang, Y.G.; Wilkins, K.J.; Hu, X.; Street, J.M.; Alvarez-Prats, A.; Mullick, A.E.; et al. Antagonism of Scavenger Receptor CD36 by 5A Peptide Prevents Chronic Kidney Disease Progression in Mice Independent of Blood Pressure Regulation. Kidney Int. 2016, 89, 809–822.
- Okamura, D.M.; Pennathur, S.; Pasichnyk, K.; López-Guisa, J.M.; Collins, S.; Febbraio, M.; Heinecke, J.; Eddy, A.A. CD36 Regulates Oxidative Stress and Inflammation in Hypercholesterolemic CKD. J. Am. Soc. Nephrol. 2009, 20, 495–505.
- Ceja-Galicia, Z.A.; García-Arroyo, F.E.; Aparicio-Trejo, O.E.; El-Hafidi, M.; Gonzaga-Sánchez, G.; León-Contreras, J.C.; Hernández-Pando, R.; Guevara-Cruz, M.; Tovar, A.R.; Rojas-Morales, P.; et al. Therapeutic Effect of Curcumin on 5/6Nx Hypertriglyceridemia: Association with the Improvement of Renal Mitochondrial β-Oxidation and Lipid Metabolism in Kidney and Liver. Antioxidants 2022, 11, 2195.
- Ly, L.D.; Xu, S.; Choi, S.-K.; Ha, C.-M.; Thoudam, T.; Cha, S.-K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.-K.; Park, K.-S. Oxidative Stress and Calcium Dysregulation by Palmitate in Type 2 Diabetes. Exp. Mol. Med. 2017, 49, e291.
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty Acid–Induced NLRP3-ASC Inflammasome Activation Interferes with Insulin Signaling. Nat. Immunol. 2011, 12, 408–415.
- Aparicio-Trejo, O.E.; Rojas-Morales, P.; Avila-Rojas, S.H.; León-Contreras, J.C.; Hernández-Pando, R.; Jiménez-Uribe, A.P.; Prieto-Carrasco, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J.; Tapia, E. Temporal Alterations in Mitochondrial β-Oxidation and Oxidative Stress Aggravate Chronic Kidney Disease Development in 5/6 Nephrectomy Induced Renal Damage. Int. J. Mol. Sci. 2020, 21, 6512.
- Aparicio-Trejo, O.E.; Avila-Rojas, S.H.; Tapia, E.; Rojas-Morales, P.; León-Contreras, J.C.; Martínez-Klimova, E.; Hernández-Pando, R.; Sánchez- Lozada, L.G.; Pedraza-Chaverri, J. Chronic Impairment of Mitochondrial Bioenergetics and β-Oxidation Promotes Experimental AKI-to-CKD Transition Induced by Folic Acid. Free Radic. Biol. Med. 2020, 154, 18–32.
- Feng, B.; Meng, R.; Huang, B.; Bi, Y.; Shen, S.; Zhu, D. Silymarin Protects against Renal Injury through Normalization of Lipid Metabolism and Mitochondrial Biogenesis in High Fat-Fed Mice. Free Radic. Biol. Med. 2017, 110, 240–249.
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered Renal Lipid Metabolism and Renal Lipid Accumulation in Human Diabetic Nephropathy. J. Lipid Res. 2014, 55, 561–572.
- Yang, X.; Okamura, D.M.; Lu, X.; Chen, Y.; Moorhead, J.; Varghese, Z.; Ruan, X.Z. CD36 in Chronic Kidney Disease: Novel Insights and Therapeutic Opportunities. Nat. Rev. Nephrol. 2017, 13, 769–781.
- Helal, I.; Fick-Brosnahan, G.M.; Reed-Gitomer, B.; Schrier, R.W. Glomerular Hyperfiltration: Definitions, Mechanisms and Clinical Implications. Nat. Rev. Nephrol. 2012, 8, 293–300.
- Kanbay, M.; Ertuglu, L.A.; Afsar, B.; Ozdogan, E.; Kucuksumer, Z.S.; Ortiz, A.; Covic, A.; Kuwabara, M.; Cherney, D.Z.I.; van Raalte, D.H.; et al. Renal Hyperfiltration Defined by High Estimated Glomerular Filtration Rate: A Risk Factor for Cardiovascular Disease and Mortality. Diabetes Obes. Metab. 2019, 21, 2368–2383.
- Duchen, M.R. Mitochondria and Calcium: From Cell Signalling to Cell Death. J. Physiol. 2000, 529, 57–68.
- Prieto-Carrasco, R.; García-Arroyo, F.E.; Aparicio-Trejo, O.E.; Rojas-Morales, P.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Tapia, E.; Pedraza-Chaverri, J. Progressive Reduction in Mitochondrial Mass Is Triggered by Alterations in Mitochondrial Biogenesis and Dynamics in Chronic Kidney Disease Induced by 5/6 Nephrectomy. Biology 2021, 10, 349.
- Amdur, R.L.; Feldman, H.I.; Gupta, J.; Yang, W.; Kanetsky, P.; Shlipak, M.; Rahman, M.; Lash, J.P.; Townsend, R.R.; Ojo, A.; et al. Inflammation and Progression of CKD: The CRIC Study. Clin. J. Am. Soc. Nephrol. 2016, 11, 1546–1556.
- Gupta, J.; Mitra, N.; Kanetsky, P.A.; Devaney, J.; Wing, M.R.; Reilly, M.; Shah, V.O.; Balakrishnan, V.S.; Guzman, N.J.; Girndt, M.; et al. Association between Albuminuria, Kidney Function, and Inflammatory Biomarker Profile in CKD in CRIC. Clin. J. Am. Soc. Nephrol. CJASN 2012, 7, 1938–1946.
- Jassim, A.H.; Inman, D.M.; Mitchell, C.H. Crosstalk Between Dysfunctional Mitochondria and Inflammation in Glaucomatous Neurodegeneration. Front. Pharmacol. 2021, 12, 699623.
- Huang, M.; Wei, R.; Wang, Y.; Su, T.; Li, P.; Chen, X. The Uremic Toxin Hippurate Promotes Endothelial Dysfunction via the Activation of Drp1-Mediated Mitochondrial Fission. Redox Biol. 2018, 16, 303–313.
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832.
- Jeong, S.-Y.; Seol, D.-W. The Role of Mitochondria in Apoptosis. BMB Rep. 2008, 41, 11–22.
- Sanz, A.B.; Sanchez-Niño, M.D.; Ramos, A.M.; Ortiz, A. Regulated Cell Death Pathways in Kidney Disease. Nat. Rev. Nephrol. 2023, 19, 281–299.
- Pereira, G.; Dos Santos, E.P.; Barbosa, M.R.; Arioni, S.; Santos, T.C.D.O.; Silva, D.T.D.R.E.; Agudelo, J.H.; Franco, M.P.; Fernandez, R.; Pereira, R.; et al. Thioredoxin-Interacting Protein: The Redoxissome Complex in Glomerular Lesion. Eur. J. Biol. 2022, 81, 274–280.
- Nishida, K.; Watanabe, H.; Murata, R.; Tokumaru, K.; Fujimura, R.; Oshiro, S.; Nagasaki, T.; Miyahisa, M.; Hiramoto, Y.; Nosaki, H.; et al. Recombinant Long-Acting Thioredoxin Ameliorates AKI to CKD Transition via Modulating Renal Oxidative Stress and Inflammation. Int. J. Mol. Sci. 2021, 22, 5600.
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–225.
- Whayne, T.F.; Parinandi, N.; Maulik, N. Thioredoxins in Cardiovascular Disease. Can. J. Physiol. Pharmacol. 2015, 93, 903–911.
- Dikalova, A.E.; Itani, H.A.; Nazarewicz, R.R.; McMaster, W.G.; Flynn, C.R.; Uzhachenko, R.; Fessel, J.P.; Gamboa, J.L.; Harrison, D.G.; Dikalov, S.I. Sirt3 Impairment and SOD2 Hyperacetylation in Vascular Oxidative Stress and Hypertension. Circ. Res. 2017, 121, 564–574.
- Ho, H.-J.; Shirakawa, H. Oxidative Stress and Mitochondrial Dysfunction in Chronic Kidney Disease. Cells 2022, 12, 88.
- Hu, C.; Zhang, H.; Qiao, Z.; Wang, Y.; Zhang, P.; Yang, D. Loss of Thioredoxin 2 Alters Mitochondrial Respiratory Function and Induces Cardiomyocyte Hypertrophy. Exp. Cell Res. 2018, 372, 61–72.
- Darwesh, A.M.; Jamieson, K.L.; Wang, C.; Samokhvalov, V.; Seubert, J.M. Cardioprotective Effects of CYP-Derived Epoxy Metabolites of Docosahexaenoic Acid Involve Limiting NLRP3 Inflammasome Activation. Can. J. Physiol. Pharmacol. 2019, 97, 544–556.
- Shateri, H.; Manafi, B.; Tayebinia, H.; Karimi, J.; Khodadadi, I. Imbalance in Thioredoxin System Activates NLRP3 Inflammasome Pathway in Epicardial Adipose Tissue of Patients with Coronary Artery Disease. Mol. Biol. Rep. 2021, 48, 1181–1191.
- Bugyei-Twum, A.; Abadeh, A.; Thai, K.; Zhang, Y.; Mitchell, M.; Kabir, G.; Connelly, K.A. Suppression of NLRP3 Inflammasome Activation Ameliorates Chronic Kidney Disease-Induced Cardiac Fibrosis and Diastolic Dysfunction. Sci. Rep. 2016, 6, 39551.
- Anders, H.-J.; Muruve, D.A. The Inflammasomes in Kidney Disease. J. Am. Soc. Nephrol. 2011, 22, 1007–1018.
- Aparicio-Trejo, O.E.; Aranda-Rivera, A.K.; Osorio-Alonso, H.; Martínez-Klimova, E.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J.; Tapia, E. Extracellular Vesicles in Redox Signaling and Metabolic Regulation in Chronic Kidney Disease. Antioxidants 2022, 11, 356.
- Liu, R.-M.; Desai, L.P. Reciprocal Regulation of TGF-β and Reactive Oxygen Species: A Perverse Cycle for Fibrosis. Redox Biol. 2015, 6, 565–577.
- Song, B.; Estrada, K.D.; Lyons, K.M. Smad Signaling in Skeletal Development and Regeneration. Cytokine Growth Factor Rev. 2009, 20, 379–388.
- Patel, B.; Ismahil, M.A.; Hamid, T.; Bansal, S.S.; Prabhu, S.D. Mononuclear Phagocytes Are Dispensable for Cardiac Remodeling in Established Pressure-Overload Heart Failure. PLoS ONE 2017, 12, e0170781.
- Gullestad, L.; Ueland, T.; Vinge, L.E.; Finsen, A.; Yndestad, A.; Aukrust, P. Inflammatory Cytokines in Heart Failure: Mediators and Markers. Cardiology 2012, 122, 23–35.
- Korn, S.H.; Wouters, E.F.M.; Vos, N.; Janssen-Heininger, Y.M.W. Cytokine-Induced Activation of Nuclear Factor-κB Is Inhibited by Hydrogen Peroxide through Oxidative Inactivation of IκB Kinase. J. Biol. Chem. 2001, 276, 35693–35700.
- An, N.; Gao, Y.; Si, Z.; Zhang, H.; Wang, L.; Tian, C.; Yuan, M.; Yang, X.; Li, X.; Shang, H.; et al. Regulatory Mechanisms of the NLRP3 Inflammasome, a Novel Immune-Inflammatory Marker in Cardiovascular Diseases. Front. Immunol. 2019, 10, 1592.
- Mavrogonatou, E.; Konstantinou, A.; Kletsas, D. Long-Term Exposure to TNF-α Leads Human Skin Fibroblasts to a P38 MAPK- and ROS-Mediated Premature Senescence. Biogerontology 2018, 19, 237–249.
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.-Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial Reactive Oxygen Species Promote Production of Proinflammatory Cytokines and Are Elevated in TNFR1-Associated Periodic Syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533.
- Kouri, V.-P.; Olkkonen, J.; Nurmi, K.; Peled, N.; Ainola, M.; Mandelin, J.; Nordström, D.C.; Eklund, K.K. IL-17A and TNF Synergistically Drive Expression of Proinflammatory Mediators in Synovial Fibroblasts via IκBζ-Dependent Induction of ELF3. Rheumatology 2022, 62, 872–885.
- Liu, D.; Zeng, X.; Li, X.; Mehta, J.L.; Wang, X. Role of NLRP3 Inflammasome in the Pathogenesis of Cardiovascular Diseases. Basic Res. Cardiol. 2018, 113, 5.
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating Mitochondrial DAMPs Cause Inflammatory Responses to Injury. Nature 2010, 464, 104–107.
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414.
- Sameer, A.S.; Nissar, S. Toll-Like Receptors (TLRs): Structure, Functions, Signaling, and Role of Their Polymorphisms in Colorectal Cancer Susceptibility. BioMed Res. Int. 2021, 2021, 1157023.
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801.
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like Receptors Activation, Signaling, and Targeting: An Overview. Bull. Natl. Res. Cent. 2019, 43, 187.
- Piccinini, A.M.; Midwood, K.S. DAMPening Inflammation by Modulating TLR Signalling. Mediat. Inflamm. 2010, 2010, 672395.
- Bao, W.; Xia, H.; Liang, Y.; Ye, Y.; Lu, Y.; Xu, X.; Duan, A.; He, J.; Chen, Z.; Wu, Y.; et al. Toll-like Receptor 9 Can Be Activated by Endogenous Mitochondrial DNA to Induce Podocyte Apoptosis. Sci. Rep. 2016, 6, 22579.
- Zhang, Y.; Peng, T.; Zhu, H.; Zheng, X.; Zhang, X.; Jiang, N.; Cheng, X.; Lai, X.; Shunnar, A.; Singh, M.; et al. Prevention of Hyperglycemia-Induced Myocardial Apoptosis by Gene Silencing of Toll-like Receptor-4. J. Transl. Med. 2010, 8, 133.
- Garibotto, G.; Carta, A.; Picciotto, D.; Viazzi, F.; Verzola, D. Toll-like Receptor-4 Signaling Mediates Inflammation and Tissue Injury in Diabetic Nephropathy. J. Nephrol. 2017, 30, 719–727.
- Verzola, D.; Bonanni, A.; Sofia, A.; Montecucco, F.; D’Amato, E.; Cademartori, V.; Parodi, E.L.; Viazzi, F.; Venturelli, C.; Brunori, G.; et al. Toll-like Receptor 4 Signalling Mediates Inflammation in Skeletal Muscle of Patients with Chronic Kidney Disease: Toll-like Receptor 4 in Muscle of Chronic Kidney Disease Patients. J. Cachexia Sarcopenia Muscle 2017, 8, 131–144.
- Anders, H.-J.; Suarez-Alvarez, B.; Grigorescu, M.; Foresto-Neto, O.; Steiger, S.; Desai, J.; Marschner, J.A.; Honarpisheh, M.; Shi, C.; Jordan, J.; et al. The Macrophage Phenotype and Inflammasome Component NLRP3 Contributes to Nephrocalcinosis-Related Chronic Kidney Disease Independent from IL-1–Mediated Tissue Injury. Kidney Int. 2018, 93, 656–669.
- Trentin-Sonoda, M.; Da Silva, R.C.; Kmit, F.V.; Abrahão, M.V.; Cahli, G.M.; Brasil, G.V.; Muzi-Filho, H.; Silva, P.A.; Tovar-Moll, F.F.; Vieyra, A.; et al. Knockout of Toll-Like Receptors 2 and 4 Prevents Renal Ischemia-Reperfusion-Induced Cardiac Hypertrophy in Mice. PLoS ONE 2015, 10, e0139350.
- Beere, H.M.; Wolf, B.B.; Cain, K.; Mosser, D.D.; Mahboubi, A.; Kuwana, T.; Tailor, P.; Morimoto, R.I.; Cohen, G.M.; Green, D.R. Heat-Shock Protein 70 Inhibits Apoptosis by Preventing Recruitment of Procaspase-9 to the Apaf-1 Apoptosome. Nat. Cell Biol. 2000, 2, 469–475.
- Vega, V.L.; Rodríguez-Silva, M.; Frey, T.; Gehrmann, M.; Diaz, J.C.; Steinem, C.; Multhoff, G.; Arispe, N.; De Maio, A. Hsp70 Translocates into the Plasma Membrane after Stress and Is Released into the Extracellular Environment in a Membrane-Associated Form That Activates Macrophages. J. Immunol. 2008, 180, 4299–4307.
- Triantafilou, M.; Triantafilou, K. Heat-Shock Protein 70 and Heat-Shock Protein 90 Associate with Toll-like Receptor 4 in Response to Bacterial Lipopolysaccharide. Biochem. Soc. Trans. 2004, 32, 636–639.
- Kornej, J.; Reinhardt, C.; Kosiuk, J.; Arya, A.; Hindricks, G.; Adams, V.; Husser, D.; Bollmann, A. Response of Circulating Heat Shock Protein 70 and Anti-Heat Shock Protein 70 Antibodies to Catheter Ablation of Atrial Fibrillation. J. Transl. Med. 2013, 11, 49.
- Johnson, G.L.; Lapadat, R. Mitogen-Activated Protein Kinase Pathways Mediated by ERK, JNK, and P38 Protein Kinases. Science 2002, 298, 1911–1912.
- Ries, M.; Schuster, P.; Thomann, S.; Donhauser, N.; Vollmer, J.; Schmidt, B. Identification of Novel Oligonucleotides from Mitochondrial DNA That Spontaneously Induce Plasmacytoid Dendritic Cell Activation. J. Leukoc. Biol. 2013, 94, 123–135.
- Brinkmann, M.M.; Spooner, E.; Hoebe, K.; Beutler, B.; Ploegh, H.L.; Kim, Y.-M. The Interaction between the ER Membrane Protein UNC93B and TLR3, 7, and 9 Is Crucial for TLR Signaling. J. Cell Biol. 2007, 177, 265–275.
- Fukui, R.; Saitoh, S.-I.; Kanno, A.; Onji, M.; Shibata, T.; Ito, A.; Onji, M.; Matsumoto, M.; Akira, S.; Yoshida, N.; et al. Unc93B1 Restricts Systemic Lethal Inflammation by Orchestrating Toll-like Receptor 7 and 9 Trafficking. Immunity 2011, 35, 69–81.
- Sasai, M.; Linehan, M.M.; Iwasaki, A. Bifurcation of Toll-Like Receptor 9 Signaling by Adaptor Protein 3. Science 2010, 329, 1530–1534.
- Ewald, S.E.; Lee, B.L.; Lau, L.; Wickliffe, K.E.; Shi, G.-P.; Chapman, H.A.; Barton, G.M. The Ectodomain of Toll-like Receptor 9 Is Cleaved to Generate a Functional Receptor. Nature 2008, 456, 658–662.
- Han, S.J.; Li, H.; Kim, M.; Shlomchik, M.J.; Lee, H.T. Kidney Proximal Tubular TLR9 Exacerbates Ischemic Acute Kidney Injury. J. Immunol. 2018, 201, 1073–1085.
- Murao, A.; Aziz, M.; Wang, H.; Brenner, M.; Wang, P. Release Mechanisms of Major DAMPs. Apoptosis 2021, 26, 152–162.
- Bliksøen, M.; Mariero, L.H.; Torp, M.K.; Baysa, A.; Ytrehus, K.; Haugen, F.; Seljeflot, I.; Vaage, J.; Valen, G.; Stensløkken, K.-O. Extracellular mtDNA Activates NF-κB via Toll-like Receptor 9 and Induces Cell Death in Cardiomyocytes. Basic Res. Cardiol. 2016, 111, 42.
- Lv, L.-L.; Feng, Y.; Tang, T.-T.; Liu, B.-C. New Insight into the Role of Extracellular Vesicles in Kidney Disease. J. Cell. Mol. Med. 2019, 23, 731–739.
- Karpman, D.; Ståhl, A.; Arvidsson, I. Extracellular Vesicles in Renal Disease. Nat. Rev. Nephrol. 2017, 13, 545–562.
- Battistelli, M.; Falcieri, E. Apoptotic Bodies: Particular Extracellular Vesicles Involved in Intercellular Communication. Biology 2020, 9, 21.
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132.
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215.
- Zuo, Z.; Jing, K.; Wu, H.; Wang, S.; Ye, L.; Li, Z.; Yang, C.; Pan, Q.; Liu, W.J.; Liu, H.F. Mechanisms and Functions of Mitophagy and Potential Roles in Renal Disease. Front. Physiol. 2020, 11, 935.
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230.
- Mohammad, N.S.; Nazli, R.; Zafar, H.; Fatima, S. Effects of lipid based Multiple Micronutrients Supplement on the birth outcome of underweight pre-eclamptic women: A randomized clinical trial. Pak. J. Med. Sci. 2022, 38, 219–226.
- Khan, S.; Basu, S.; Raj, D.; Lahiri, A. Role of Mitochondria in Regulating Immune Response during Bacterial Infection. Int. Rev. Cell Mol. Biol. 2023, 374, 159–200.
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The Adaptor MAVS Promotes NLRP3 Mitochondrial Localization and Inflammasome Activation. Cell 2013, 153, 348–361.
- Aranda-Rivera, A.K.; Srivastava, A.; Cruz-Gregorio, A.; Pedraza-Chaverri, J.; Mulay, S.R.; Scholze, A. Involvement of Inflammasome Components in Kidney Disease. Antioxidants 2022, 11, 246.
- Park, S.; Juliana, C.; Hong, S.; Datta, P.; Hwang, I.; Fernandes-Alnemri, T.; Yu, J.-W.; Alnemri, E.S. The Mitochondrial Antiviral Protein MAVS Associates with NLRP3 and Regulates Its Inflammasome Activity. J. Immunol. 2013, 191, 4358–4366.
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host Cell Death and Inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109.
- Zheng, X.; Chen, W.; Gong, F.; Chen, Y.; Chen, E. The Role and Mechanism of Pyroptosis and Potential Therapeutic Targets in Sepsis: A Review. Front. Immunol. 2021, 12, 711939.
- Chen, X.; He, W.; Hu, L.; Li, J.; Fang, Y.; Wang, X.; Xu, X.; Wang, Z.; Huang, K.; Han, J. Pyroptosis Is Driven by Non-Selective Gasdermin-D Pore and Its Morphology Is Different from MLKL Channel-Mediated Necroptosis. Cell Res. 2016, 26, 1007–1020.
- Gurung, P.; Lukens, J.R.; Kanneganti, T.-D. Mitochondria: Diversity in the Regulation of the NLRP3 Inflammasome. Trends Mol. Med. 2015, 21, 193–201.
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS–STING Pathway as a Therapeutic Target in Inflammatory Diseases. Nat. Rev. Immunol. 2021, 21, 548–569.
- Ma, X.M.; Geng, K.; Law, B.Y.-K.; Wang, P.; Pu, Y.L.; Chen, Q.; Xu, H.W.; Tan, X.Z.; Jiang, Z.Z.; Xu, Y. Lipotoxicity-Induced mtDNA Release Promotes Diabetic Cardiomyopathy by Activating the cGAS-STING Pathway in Obesity-Related Diabetes. Cell Biol. Toxicol. 2022, 39, 277–299.
- Zang, N.; Cui, C.; Guo, X.; Song, J.; Hu, H.; Yang, M.; Xu, M.; Wang, L.; Hou, X.; He, Q.; et al. cGAS-STING Activation Contributes to Podocyte Injury in Diabetic Kidney Disease. iScience 2022, 25, 105145.
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791.
- Dobbs, N.; Burnaevskiy, N.; Chen, D.; Gonugunta, V.K.; Alto, N.M.; Yan, N. STING Activation by Translocation from the ER Is Associated with Infection and Autoinflammatory Disease. Cell Host Microbe 2015, 18, 157–168.
- Ishikawa, H.; Barber, G.N. STING Is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature 2008, 455, 674–678.
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING Regulates Intracellular DNA-Mediated, Type I Interferon-Dependent Innate Immunity. Nature 2009, 461, 788–792.
- Chung, K.W.; Dhillon, P.; Huang, S.; Sheng, X.; Shrestha, R.; Qiu, C.; Kaufman, B.A.; Park, J.; Pei, L.; Baur, J.; et al. Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab. 2019, 30, 784–799.e5.
- Mitrofanova, A.; Fontanella, A.; Tolerico, M.; Mallela, S.; David, J.M.; Zuo, Y.; Boulina, M.; Kim, J.-J.; Santos, J.; Ge, M.; et al. Activation of Stimulator of IFN Genes (STING) Causes Proteinuria and Contributes to Glomerular Diseases. J. Am. Soc. Nephrol. 2022, 33, 2153–2173.
- Nishimoto, S.; Sata, M.; Fukuda, D. Expanding Role of Deoxyribonucleic Acid-Sensing Mechanism in the Development of Lifestyle-Related Diseases. Front. Cardiovasc. Med. 2022, 9, 881181.
- Gong, W.; Lu, L.; Zhou, Y.; Liu, J.; Ma, H.; Fu, L.; Huang, S.; Zhang, Y.; Zhang, A.; Jia, Z. The Novel STING Antagonist H151 Ameliorates Cisplatin-Induced Acute Kidney Injury and Mitochondrial Dysfunction. Am. J. Physiol.-Ren. Physiol. 2021, 320, F608–F616.
- Yu, C.-H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e18.
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond–Mitochondrial Performance in Apoptosis. FEBS J. 2018, 285, 416–431.
- Renault, T.T.; Chipuk, J.E. Death upon a Kiss: Mitochondrial Outer Membrane Composition and Organelle Communication Govern Sensitivity to BAK/BAX-Dependent Apoptosis. Chem. Biol. 2014, 21, 114–123.
- Maekawa, H.; Inoue, T.; Ouchi, H.; Jao, T.-M.; Inoue, R.; Nishi, H.; Fujii, R.; Ishidate, F.; Tanaka, T.; Tanaka, Y.; et al. Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep. 2019, 29, 1261–1273.e6.
- Khedr, S.; Dissanayake, L.V.; Palygin, O.; Staruschenko, A. Potential Role of cGAS-STING Pathway in the Induction of Diabetic Kidney Disease. FASEB J. 2020, 34, 1.
- Aarreberg, L.D.; Esser-Nobis, K.; Driscoll, C.; Shuvarikov, A.; Roby, J.A.; Gale, M. Interleukin-1β Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol. Cell 2019, 74, 801–815.e6.
- Akazawa, Y.; Nakashima, R.; Matsuda, K.; Okamaoto, K.; Hirano, R.; Kawasaki, H.; Miuma, S.; Miyaaki, H.; Malhi, H.; Abiru, S.; et al. Detection of DNA Damage Response in Nonalcoholic Fatty Liver Disease via P53-Binding Protein 1 Nuclear Expression. Mod. Pathol. 2019, 32, 997–1007.
- Bhandari, S. Risk Factors and Metabolic Mechanisms in the Pathogenesis of Uraemic Cardiac Disease. Front. Biosci. 2011, 16, 1364–1387.
- Charo, I.F.; Ransohoff, R.M. The Many Roles of Chemokines and Chemokine Receptors in Inflammation. N. Engl. J. Med. 2006, 354, 610–621.
- Bandow, K.; Kusuyama, J.; Shamoto, M.; Kakimoto, K.; Ohnishi, T.; Matsuguchi, T. LPS-Induced Chemokine Expression in Both MyD88-Dependent and -Independent Manners Is Regulated by Cot/Tpl2-ERK Axis in Macrophages. FEBS Lett. 2012, 586, 1540–1546.
- Nomiyama, H.; Osada, N.; Yoshie, O. Systematic Classification of Vertebrate Chemokines Based on Conserved Synteny and Evolutionary History. Genes Cells 2013, 18, 1–16.
- Chang, T.-T.; Chen, C.; Chen, J.-W. CCL7 as a Novel Inflammatory Mediator in Cardiovascular Disease, Diabetes Mellitus, and Kidney Disease. Cardiovasc. Diabetol. 2022, 21, 185.
- DeVries, M.E.; Kelvin, A.A.; Xu, L.; Ran, L.; Robinson, J.; Kelvin, D.J. Defining the Origins and Evolution of the Chemokine/Chemokine Receptor System. J. Immunol. 2006, 176, 401–415.
- Chang, T.-T.; Chen, J.-W. The Role of Chemokines and Chemokine Receptors in Diabetic Nephropathy. Int. J. Mol. Sci. 2020, 21, 3172.
- Richmond, A. NF-κB, Chemokine Gene Transcription and Tumour Growth. Nat. Rev. Immunol. 2002, 2, 664–674.
- Segerer, S.; Nelson, P.J.; Schlöndorff, D. Chemokines, Chemokine Receptors, and Renal Disease: From Basic Science to Pathophysiologic and Therapeutic Studies. J. Am. Soc. Nephrol. 2000, 11, 152–176.
- Chung, A.C.K.; Lan, H.Y. Chemokines in Renal Injury. J. Am. Soc. Nephrol. 2011, 22, 802–809.
- Galliera, E.; Corsi, M.; Bonecchi, R.; Locati, M.; Mantovani, A. Chemokines as Pharmacological Targets. Mini-Rev. Med. Chem. 2008, 8, 638–646.
- Vielhauer, V.; Anders, H.-J. Chemokines and Chemokine Receptors as Therapeutic Targets in Chronic Kidney Disease. Front. Biosci. 2009, S1, 1–12.
- Miao, M.; De Clercq, E.; Li, G. Clinical Significance of Chemokine Receptor Antagonists. Expert Opin. Drug Metab. Toxicol. 2020, 16, 11–30.
- Tang, S.C.W.; Lai, K.N. The Pathogenic Role of the Renal Proximal Tubular Cell in Diabetic Nephropathy. Nephrol. Dial. Transplant. 2012, 27, 3049–3056.
- Anders, H.-J.; Ryu, M. Renal Microenvironments and Macrophage Phenotypes Determine Progression or Resolution of Renal Inflammation and Fibrosis. Kidney Int. 2011, 80, 915–925.
- Weidenbusch, M.; Anders, H.-J. Tissue Microenvironments Define and Get Reinforced by Macrophage Phenotypes in Homeostasis or during Inflammation, Repair and Fibrosis. J. Innate Immun. 2012, 4, 463–477.
- Wang, Y.; Rangan, G.K.; Tay, Y.-C.; Wang, Y.; Harris, D.C.H. Induction of Monocyte Chemoattractant Protein-1 by Albumin Is Mediated by Nuclear Factor κB in Proximal Tubule Cells. J. Am. Soc. Nephrol. 1999, 10, 1204–1213.
- Zoja, C.; Donadelli, R.; Colleoni, S.; Figliuzzi, M.; Bonazzola, S.; Morigi, M.; Remuzzi, G. Protein Overload Stimulates RANTES Production by Proximal Tubular Cells Depending on NF-kB Activation. Kidney Int. 1998, 53, 1608–1615.
- Huang, X.R.; Chung, A.C.K.; Zhou, L.; Wang, X.J.; Lan, H.Y. Latent TGF-β1 Protects against Crescentic Glomerulonephritis. J. Am. Soc. Nephrol. JASN 2008, 19, 233–242.
- Chevalier, R.L.; Forbes, M.S.; Thornhill, B.A. Ureteral Obstruction as a Model of Renal Interstitial Fibrosis and Obstructive Nephropathy. Kidney Int. 2009, 75, 1145–1152.
- Chung, A.C.K.; Huang, X.R.; Zhou, L.; Heuchel, R.; Lai, K.N.; Lan, H.Y. Disruption of the Smad7 Gene Promotes Renal Fibrosis and Inflammation in Unilateral Ureteral Obstruction (UUO) in Mice. Nephrol. Dial. Transplant. 2009, 24, 1443–1454.
- Islam, S.A.; Chang, D.S.; Colvin, R.A.; Byrne, M.H.; McCully, M.L.; Moser, B.; Lira, S.A.; Charo, I.F.; Luster, A.D. Mouse CCL8, a CCR8 Agonist, Promotes Atopic Dermatitis by Recruiting IL-5+ TH2 Cells. Nat. Immunol. 2011, 12, 167–177.
- Akchurin, O.M.; Kaskel, F. Update on Inflammation in Chronic Kidney Disease. Blood Purif. 2015, 39, 84–92.
- Lee, Y.; Lee, J.; Park, M.; Seo, A.; Kim, K.H.; Kim, S.; Kang, M.; Kang, E.; Yoo, K.D.; Lee, S.; et al. Inflammatory Chemokine (C-C Motif) Ligand 8 Inhibition Ameliorates Peritoneal Fibrosis. FASEB J. 2023, 37, e22632.
- Luster, A.D.; Ravetch, J.V. Biochemical Characterization of a Gamma Interferon-Inducible Cytokine (IP-10). J. Exp. Med. 1987, 166, 1084–1097.
- Dyer, K.D.; Percopo, C.M.; Fischer, E.R.; Gabryszewski, S.J.; Rosenberg, H.F. Pneumoviruses Infect Eosinophils and Elicit MyD88-Dependent Release of Chemoattractant Cytokines and Interleukin-6. Blood 2009, 114, 2649–2656.
- Panzer, U.; Steinmetz, O.M.; Reinking, R.R.; Meyer, T.N.; Fehr, S.; Schneider, A.; Zahner, G.; Wolf, G.; Helmchen, U.; Schaerli, P.; et al. Compartment-Specific Expression and Function of the Chemokine IP-10/CXCL10 in a Model of Renal Endothelial Microvascular Injury. J. Am. Soc. Nephrol. 2006, 17, 454–464.