Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Tadayuki Komori and Version 2 by Lindsay Dong.
Studies point to the involvement of endolysosomal defects in parkinson’s disease (PD). The endolysosomal system, which tightly controls a flow of endocytosed vesicles targeted either for degradation or recycling, is regulated by a number of Rab GTPases. Their associations with leucine-rich repeat kinase 2 (LRRK2), a major causative and risk protein of PD, has also been one of the hot topics in the field.
- LRRK2
- Rab
- endolysosome
- intracellular transport
- Parkinson’s disease
1. Introduction
Since its discovery as the protein responsible for Parkinson’s disease (PD) in the PARK8 locus in 2004 [1][2][1,2], leucine-rich repeat kinase 2 (LRRK2) has been one of the main focus molecules associated with this neurodegenerative disease. The physiological roles of LRRK2 have been linked to a myriad of cellular processes, such as several types of autophagy including macroautophagy and mitophagy [3][4][3,4], endocytosis and intracellular transport involving the trans-Golgi network (TGN) and other organelles [5][6][7][8][5,6,7,8], the regulation of microtubules [5][6][5,6], interaction with bacterial pathogens [7], regulation of lysosomal homeostasis [8], and much more.
The pathogenic features of LRRK2 have also been studied by analyzing mutations associated with PD, and most, if not all, mutations point towards a similar effect: augmentation of substrate phosphorylation [8][9][8,9]. Albeit these findings, the exact mechanism of how defects in LRRK2 lead to PD has been elusive for nearly a score of years now. An auspicious approach to this enigma would be its link to the endolysosomal system and their regulators, Rab GTPases, as a considerable number of findings indicate connections between these [10].
Rab GTPases bind to membranes and utilize their affinity change via their guanine nucleotide binding status to form specific functional domains on their corresponding organelle membranes [11], hence called the master regulators of intracellular vesicular traffic. Some of the first reports that related Rab GTPases to PD were around 20 years ago, when α-synuclein was reported to interact with several Rab GTPases [12] or α-synuclein-induced neuronal loss was rescued by overexpression of Rab1 [13].
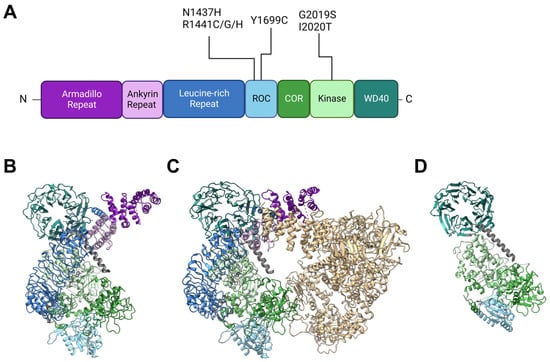
2. Insights from Genetic and Structural Studies of LRRK2
Back in 2002, Funayama et al. reported a family of inherited PD with an unknown causative gene locus, PARK8 [14][20]. Two years later, two independent groups identified the responsible gene as LRRK2 [1][2][1,2]. Since then, genetics have revealed at least seven causative mutations in this gene product: N1437H [15][21], R1441C [1][16][1,22], R1441G [16][22], R1441H [16][22], Y1699C [1], G2019S [17][23], I2020T [1], and numerous other rare variants with unclear causality [18][24]. The gene product LRRK2 is a 2527-amino-acid multidomain kinase, and the PD-associated mutations lie in the ROC (Ras of complex), COR (C-terminal of ROC), and kinase domains (Figure 1). The ROC domain is a GTP-binding domain that regulates the kinase activity in an intramolecular fashion [19][20][25,26], although its GTP-binding state (not GTP-binding capacity) may not be required for its kinase activity [21][27]. Other domains in LRRK2 have no PD-associated mutants allocated to them but are also of importance when considering intermolecular interactions. Armadillo repeats (ARM), ankyrin repeats (ANK), and leucine-rich repeats (LRR) are relatively abundant motifs that form various sizes of scaffolds for protein interaction [22][23][24][33,34,35], whereas WD40 domains are beta-propellers that may interact with DNA as well as proteins [25][26][36,37]. The detailed primary structure is depicted in Figure 1A. It might also be worth noting that a portion of endogenous LRRK2 in macrophages is found cleaved at the ANK-LRR interdomain region to produce a C-terminal fragment including the kinase region [27][38]. Although this fragment may be nonfunctional because the N-terminal membrane-interacting region is lacking, it may also act as dominant negative as it can heterodimerize with full-length LRRK2 [27][38].Figure 1. Structures of LRRK2 and PD-associated mutations. (A) Primary structure of LRRK2 and the location of reported PD-associated mutations. ROC: Ras of complex domain, COR: C terminal of ROC domain, WD40: WD40 domain. Domain lengths and borders are based on [28][39]. (B) Monomeric structure of full-length LRRK2 [29][30] (PDB ID: 7HLT). (C) Homodimeric structure of full-length LRRK2 [29][30] (PDB ID: 7HLW). (D) Protomer of filamentous LRRK2 C-terminal half (RCKW) on microtubules [30][40] (PDB ID: 6VNO). All structures are aligned so that the WD40 domain lies at the top left. Colorization of structures is the same for (A–D).
LRRK2 is observed in many conformations in vitro or in cells, with structural models from monomer [29][30] to homodimer [29][31][32][30,41,42] to filamentous [30][33][40,43] and even heteromultimer [34][44] reported (Figure 1B–D). The classical model of LRRK2 is the homodimer model, based on biochemical analysis and crystal structures of ROC domains [35][36][45,46]. This model was further confirmed via other methods such as cross-linking and negative stain [37][47], and further with full-length LRRK2 [29][30] (Figure 1B).
The monomeric model was first proposed based on biochemical analysis of full-length LRRK2 [38][50] but was not confirmed until some years ago when the structure of full-length LRRK2 was finally determined via cryoelectron microscopy (cryo-EM) [29][30] (Figure 1C).
The filamentous LRRK2 model is based on observations of overexpressed PD-associated mutants, which are mostly cytosolic but often form filaments along microtubules [39][51]. Reports that analyzed this form of LRRK2 bound to microtubules via cryo-EM or cryoelectron tomography (cryo-ET) showed multimer formation via interactions through the WD40 domain with the help of microtubule filaments [30][33][40,43] (a protomer of the filament is shown in Figure 1D).
3. Rab GTPases and LRRK2
3.1. Rab GTPases and the Endolysosomal System
Rab GTPases are proteins that form a subfamily of the Ras superfamily and are capable of binding cellular membranes via the C-terminal prenylation. Like other small GTPases, they switch their activity, which influences their affinity to specific proteins called effectors by changing their binding state with either GTP (active) or GDP (inactive) with the help of their specific guanine nucleotide exchange factors (GEF) or GTPase-activating protein (GAP). Their functions are associated with a wide variety of intracellular trafficking, ranging from cellular secretory pathways to intracellular degradation pathways involving the endolysosomal system. The endolysosomal system is part of an intracellular flow of enveloped membrane organelles and, apart from its function in autophagy, serves as a sorting site for substances incorporated by endocytosis. These substances include extracellular materials as well as membrane proteins and cellular membranes themselves. These substances are then either guided towards degradation by lysosomes, returned (or “recycled”) to the plasma membrane, or routed to the TGN. This pathway is known to regulate basic steps of cellular processes such as signaling, adhesion, immunity, nutrient uptake, organelle homeostasis, membrane protein turnover, and much more (reviewed in [40][41][42][43][44][45][57,58,59,60,61,62]). This route to degradation can be broken up into several parts, with more than one Rab GTPase regulating the routing or maturation of each vesicle. Endocytosed materials are first retained in the early endosome, where Rab5 controls the maturation of the vesicle and other Rab GTPases, such as Rab11, regulate re-routing from the early endosome to other compartments, in this case to the recycling endosomes, another part of the endolysosomal system, and ultimately to the plasma membrane [46][47][63,64].3.2. LRRK2 and Substrate Rab GTPases
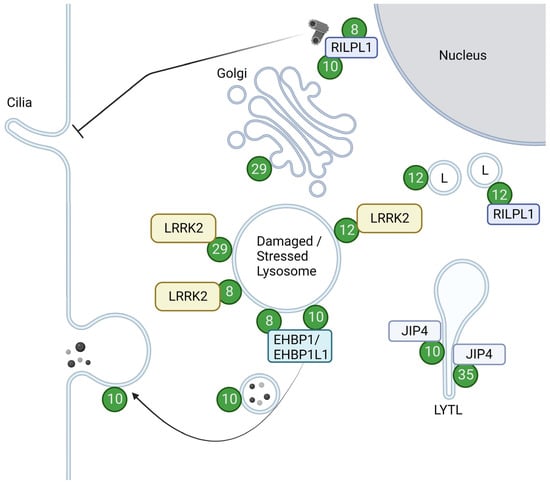
As a kinase, LRRK2 phosphorylates a subset of Rab GTPases, e.g., Rab8 and Rab10, in cells. The overview of the Rabs with their roles upon phosphorylation is summarized in Figure 2.Figure 2. Localizations and functions of LRRK2-phosphorylated Rabs. Green circles with numbers indicate Rabs phosphorylated by LRRK2. LRRK2-phosphorylated Rab12 and Rab29 are recruited to damaged or stressed lysosomes and further activate LRRK2. LRRK2-phosphorylated Rab8 and Rab10, bound to their effector RILPL1, accumulate near centrosomes to inhibit ciliogenesis. Rab8 and Rab10 also act together with their effector EHBP1 or EHBP1L1 to counteract lysosomal inflation or facilitate lysosomal release. LRRK2-phosphorylated Rab10 and Rab35 bind to JIP4 and induce LYTL. LRRK2-phosphorylated Rab12 binds to RILPL1 and moves lysosomes to the perinuclear region. LRRK2-phosphorylated Rab29 alters the morphology of the trans-Golgi. L: Lysosomes. Figure created with BioRender.com.
3.2.1. Rab8 and Rab10
Rab8 and Rab10 are closely related Rabs, both categorized in the Rab8 subfamily [48][89] and are the most characterized Rab GTPase in the context of interaction with LRRK2.
Right after the initial report of multiple Rab phosphorylation by LRRK2 [49][14], Rab10 was found to be a very sensitive marker for assessing LRRK2 activity [50][90], followed by a quick development of a phospho-specific antibody against Rab10 [51][68]. To date, numerous studies have incorporated an assessment of this phosphorylation in their studies on LRRK2 kinase activity [18][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67][24,55,76,77,78,81,82,85,91,92,93,94,95,96,97,98,99].
Rab10 has been implicated in several modes of transport in a variety of cell types, from general exocytic pathways to neurite or cilia formation and immune responses [68][110]. LRRK2 has been shown to play a role in these functions via its kinase activity, altering the ability of Rab10 to interact with other proteins. In the context of ciliogenesis, phosphorylated Rab10 binds to RILPL1 at or near centrosomes, inhibiting ciliogenesis [55][69][70][71][72][15,71,78,79,80]. This inhibition was also brought about by the retention of Myosin Va at centrioles by the same phosphorylation of Rab10 [54][77].
Rab8, on the other hand, is well characterized as a Rab GTPase, with GEFs Rabin8 and GRAB, GAPs TBC1D30, and other TBC family proteins, and multiple effectors described in the context of anterograde trafficking, endocytic recycling and exocytosis, association with the cytoskeleton, cell shape regulation and migration, ciliogenesis, neurite growth, and much more [73][111].
The first reports on Rab8 functions date back about 30 years when Rab8 was deemed responsible for post-Golgi anterograde trafficking in epithelial and neuronal cells [74][75][113,114]. Studies on Rab8’s involvement in neurite formation immediately followed [76][115] and built the classical view of Rab8 as a controller of neurite formation via anterograde trafficking. Current knowledge on Rab8 in neurite formation includes additional upstream elements, involving various other Rabs and their effectors, such as Rab11 and Rabin8, that activate Rab8 and Rab10 for neurite outgrowth [77][116], as well as downstream elements such as Cdc42 and tuba that strictly regulate the number of axons formed per cell [78][117].
Another aspect of Rab8 is its involvement in cilia. Formation of cilia requires Rab8 activation on the centrosome by Rabin8, very much like in neurite formation, and further protein trafficking to the base of primary cilia [73][111]. Impaired receptor trafficking to cilia via Rab8 dysfunction causes various deficits in functions that require cilia, such as adipocyte differentiation, where Rab8 is responsible for the trafficking of frz2 to the base of primary cilia [79][118]. Rab10 and Rab13 might have compensatory roles in ciliogenesis as double knockouts of Rab8a and Rab8b were insufficient to cause cilial deficits [80][119].