The genetic variations in Patatin-like Phospholipase Domain-containing 3 (PNPLA3), Transmembrane 6 Superfamily Member 2 (TM6SF2), and Membrane Bound O-acyltransferase Domain-containing 7 (MBOAT7) play the principal roles in the development and progression of MAFLD. The PNPLA3 gene is associated with increased hepatic lipid levels and hepatic inflammation, hepatic fibrogenesis, and elevated risk of development of HCC
[4]. TM6SF2 is associated with hepatic steatosis and progression of liver disease, because it increases the liver TG content and decreases VLDL secretion
[5]. Variants of the MBOAT7 gene are related to alcoholic cirrhosis, but also are associated with the development and severity of MAFLD via remodeling of phospholipids
[6].
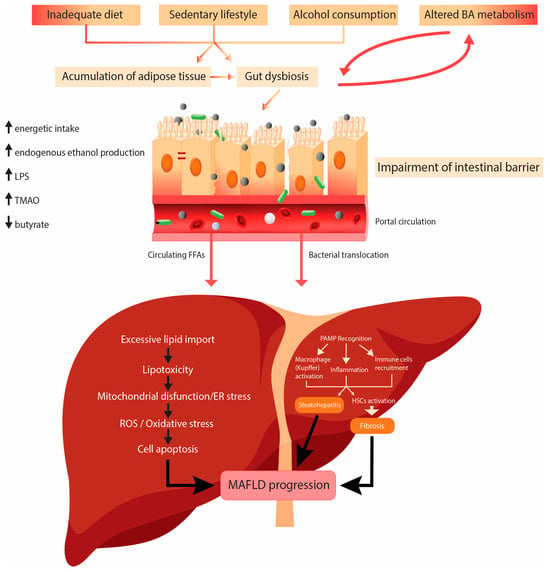
Environmental factors like high-caloric diet and decreased physical activity play a role in the pathogenesis of MAFLD.
High-fat diet (HFD) means high consumption of fat, typically known as the Western diet. High fat intake plays a significant role in the pathogenesis of MAFLD, and it leads to excessive accumulation of fat in the liver in patients with a genetic predisposition. HFD is very often associated with insulin resistance, lipid disorders, and metabolic and cardiovascular diseases, components of metabolic syndrome. The hypothesis that most likely explains the theory of progression MAFLD to nonalcoholic steatohepatitis (NASH), cirrhosis, and hepatocellular carcinoma is the double-hit hypothesis. The primary disorder contributing to steatosis is IR. IR starts hepatic de novo lipogenesis (DNL) and impaired fatty acid (FA) transport. The second hit implies mitochondrial dysfunction, endoplasmic reticulum stress, inflammation, and many other disturbances
[7].
The fatty acids (FAs) in the liver originate from dietary fat, adipose tissue lipolysis, or de novo lipogenesis (DNL) from carbohydrates or other dietary fat precursors. Inside the liver, FAs are esterified into TG and assembled into very-low-density lipoprotein (VLDL) to be secreted in the circulation, oxidized in the mitochondria (β-oxidation), or stored in lipid droplets (LDs). LDs undergo lipid hydrolysis in fasting conditions to provide FAs for β-oxidation. With chronic nutrient overload and IR in MAFLD, the input of FAs to the liver exceeds their disposal via VLDL secretion or β-oxidation
[8]. In patients with MAFLD, about 15% of liver FAs originate from the diet, 59% are derived from circulation, and 26% from DNL
[9]. IR increases DNL in the liver
[10][11][10,11]. Two main transcriptional factors are involved in DNL: sterol regulatory element binding protein 1c (SREBP1c), regulated by insulin signaling, and carbohydrate response element binding protein (ChREBP), activated by glucose uptake
[12]. The results of mice studies have shown the important role of the peroxisome proliferator-activated receptors (PPARγ) in hepatic lipogenesis
[13].
Lipotoxicity is defined as accumulation or transient generation of toxic lipids in LDs (>5% of liver weight), which may result in hepatocyte injury or death. The toxic lipids are the ones that cause cellular injury and cell death in MAFLD pathogenesis. The saturated fatty acids (SFAs) (palmitate, ceramides, lysophosphatidylcholine, and free cholesterol) are directly cytotoxic and can induce progressive liver injury. The monounsaturated free FAs, such as oleate and palmitoleate, may protect from lipotoxicity
[14]. The mechanisms of lipotoxicity involve several cellular processes in the liver, such as endoplasmic reticulum (ER) stress, mitochondrial dysfunction, oxidative stress and production of reactive oxygen species (ROS), and intestinal dysbiosis
[15].
Mitochondria, ER, and peroxisomes produce ROS in normal cellular metabolism, but 90% of cellular ROS are produced in mitochondria. An HFD increases the amount of substrate available for oxidation and the production of free radicals and damages the antioxidant defense
[16]. An imbalance between the production of ROS and the antioxidant activities leads to oxidative stress. Antioxidants with main function to eliminate free radicals involve nonenzymatic molecules such as ascorbic acid (vitamin C), α-tocopherol (vitamin E), glutathione (GSH), carotenoids, and flavonoids and the antioxidant enzymes, such as superoxide dismutase, glutathione peroxidase, and catalase. Excessive ROS production causes mitochondrial damage and activation of Kupffer cells, which then produce cytokines and cause inflammation. ROS activates hepatic stellate cells (HSCs) to produce extracellular matrix, leading to fibrosis
[8][16][17][8,16,17].
Mitochondrial dysfunction is a consequence of enormous production of ROS, but in this vicious circle, mitochondrial dysfunction is also the cause of increased production of ROS. Uncontrolled production of ROS in the mitochondria damages mitochondrial components and leads to activation of the mitochondrial quality control (MQC) system. MQC is a specific mechanism responsible for maintaining mitochondrial integrity. This system includes biogenesis, fission, fusion, and mitophagy. Enormous production of ROS can damage structural parts of mitochondria, mitochondrial membrane, DNA, and can activate apoptotic processes, leading to mitochondrial autophagy, known as mitophagy
[18][19][20][18,19,20].
A recent study in a “human-like” MAFLD rat model, which was induced by HFD-feeding for 14 weeks in association with thermoneutral housing, showed the effect of HFD on DNA damage and mitochondrial quality control mechanisms in an MAFLD. The results of this study showed that the mtDNA copy number and expression of markers involved in mitochondrial biogenesis were significantly reduced in the HFD rats group. Also, the protein levels of MFN2 (mitofusin 2), GTPase protein, which regulates the process of mitochondria, were significantly reduced in the liver of the HFD rats, but there were no significant changes in the other protein levels of fusion-optic atrophy 1 (OPA1), or in the protein levels of fission-dynamin-related protein 1 (DRP1). These results suggest a disorder of mitochondrial dynamic in MAFLD, probably more of fission. The analysis showed that, in HFD rats, the phosphorylation/activation of AMPK on Thr172 and of unc-51-like kinase 1 (ULK1) on Ser555 were both significantly increased. A significantly increased expression of AMBRA1, binding partners, and downstream effectors of ULK1 confirmed the activation of autophagy in the HFD group. A significant decrease in the expression of key proteins involved in the mitophagy flux in the liver of the HFD rats, PTEN-induced kinase 1(PINK1), the E3 ligase (PARKIN), and microtubule-associated protein 1A/1B-light chain 3B (LC3BII), indicated an impaired autophagic flux
[21].
It has been known for a long time that oxidative stress can be assessed using serum marker serum, soluble NOX2-derived peptide, and urine marker, urinary 8-iso-PGF2α. The results of the studies showed that urinary 8-iso-PGF2α and serum-soluble NOX2-derived peptide are increased in patients with nonalcoholic fatty liver. Their level is independent of obesity, diabetes, and metabolic syndrome. Levels of urinary 8-iso-PGF2α and serum-soluble NOX2-derived peptide increase with the severity of liver steatosis at ultrasound. The mentioned study indicated that the markers of oxidative stress can also be used in assessing the severity of liver steatosis
[22].
Since ER is involved in both protein and lipid homeostasis, proteotoxic and lipotoxic stress are closely related, and are likely bidirectional. Disequilibrium in the lipid bilayer induces lipotoxic ER stress, while the accumulation of unfolded or misfolded proteins in the ER lumen triggers proteotoxic ER stress. The unfolded protein sensors can be directly activated by toxic lipids. The unfolded protein response (UPR) is activated via the luminal domains of three principal transmembrane sensors: inositol-requiring enzyme (IRE)-1α, protein kinase RNA-like ER kinase (PERK), and activating transcription factor (ATF)-6α. ER is involved in protein folding and trafficking and lipid biosynthesis and trafficking. That is the reason these two types of stress are interconnected. When homeostasis is disturbed and equilibrium cannot be established, ER stress-induced apoptosis occurs
[23][24][23,24].