Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Mona Zou and Version 1 by Pascale Bomont.
Autophagy is a major degradative pathway that plays a key role in sustaining cell homeostasis, integrity, and physiological functions. Macroautophagy, which ensures the clearance of cytoplasmic components engulfed in a double-membrane autophagosome that fuses with lysosomes, is orchestrated by a complex cascade of events. Autophagy has a particularly strong impact on the nervous system, and mutations in core components cause numerous neurological diseases.
- autophagy
- regulations
- compartmentalisation
- neurodevelopment
- neurodegeneration
1. Compartmentalisation of Neuronal Autophagy
Neurons are specialised and polarised cells with three different compartments: the soma, the dendrites, and the axon. To sustain specialised functions, neuronal protein composition must be tightly regulated in each compartment, from synthesis to degradation. Moreover, given their post-mitotic nature, neurons cannot dilute toxic components through cell division and are therefore highly dependent on efficient degradative pathways. It is considered that in contrary to other cell types, neurons continuously generate autophagosomes under basal conditions. Neurons undergo compartment-specific autophagy regulation [130][1] (Figure 21). Studies have revealed that autophagosome biogenesis occurs in the distal part of the axon; maturation occurs during retrograde transport, while their content is degraded in the soma upon fusion with lysosomes. Interestingly, another site of biogenesis has been identified in the somatodendritic compartment, but their dynamics and functions are poorly understood.
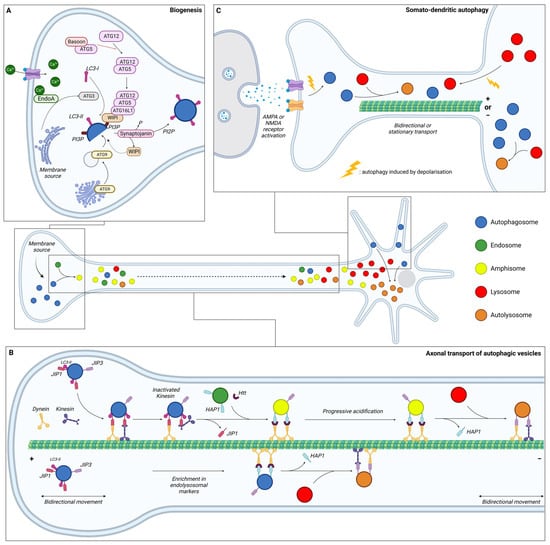
Figure 21. Schematic representation of the dynamics of neuronal autophagy. (A) Autophagosome (blue) biogenesis takes place in the distal part of the axon. (B) After their formation, autophagosomes first undergo bidirectional movement and switch to retrograde transport to move towards the soma along microtubules with the help of motor proteins (dynein and kinesin). During this transport, autophagic vesicles (AVs) mature by fusing with endosome (green) and/or lysosome (red) to generate, respectively, amphisome (yellow) and/or autolysosome (orange). In the proximity of the soma, AVs become more mature and exhibit bidirectional movement. (C) Dendritic autophagosome biogenesis is regulated by synaptic activity. Depolarisation, induced by KCl or through AMPAR/NMDAR, increases the density of AVs in dendrites and decreases displacement. Lysosomes from the soma are also recruited to dendrites after depolarisation. In somal autophagy, AVs tend to be less mobile than in axons or dendrites. This compartment is the main site of final autolysosome content degradation. Ca: calcium; EndoA: endophilin A; JIP: JNK-interacting protein; Htt: Huntingtin; HAP1: Huntingtin-associated protein 1; AMPA: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; and NMDA: N-methyl-D-aspartate.
1.1. Autophagy in the Axon
1.1.1. Biogenesis
Initial research suggested that autophagosome formation occurs in the distal part of the axons (Figure 21A). Indeed, dense-cored vesicles, formed at least in part from the reticulum, were described in the growth cone of isolated sympathetic neurons in vitro [131][2]. Identified as AVs containing cytoplasmic material, organelles were shown to be mobile through retrograde transport in the axon of peripheral neurons [132][3]. Using the GFP-LC3 probe in live imaging, ring-like structures of approximately 800 nm in diameter were found accumulated near the distal end of the axon [133][4]. These vesicles were identified as autophagosomes based on their strong LC3-positive signals and the similarity in morphology with the LC3-positive structures seen in vivo in transgenic GFP-LC3 mice [5], indicating de novo formation of autophagosomes in the distal tip.
In contrary to non-neuronal cell types, whose membranes can be of multiple origin, endoplasmic reticulum (ER) is the major source of membrane for nascent autophagosomes in neurons. More specifically, AVs emerge from specialized subdomains of the ER labelled by double-FYVE-containing protein 1 (DFCP1). As discussed in the first part of this review, autotophagosome formation is dependent on the transmembrane core protein ATG9. This lipid scramblase derives from the trans-Golgi network and mediates the translocation of phospholipids between two lipidic membranes, thus providing a phospholipid source for autophagosomal membrane expansion [134][6]. Thus, mislocalisation of ATG9A or its retention in the trans-Golgi network, as seen in HSP, reduces autophagosome formation [80,135][7][8]. Regulating exocytosis and endocytosis, ATG9 is also implicated in synaptic activity and its mislocalisation is associated with defects in activity-induced synaptic autophagy [136,137][9][10]. In C. elegans, the synaptic protein Clarinet (CLA-1L), bearing similarity to the vertebrate Rab-interacting molecule (RIM), Piccolo, and Bassoon, regulates the localisation of ATG-9 at presynaptic sites, thus promoting autophagy during neuronal activity. Indeed, Cla-1(L) mutants were found to disrupt ATG-9 sorting and to induce deficits in autophagosome formation at the synapses [138][11].
Autophagosome biogenesis in the distal tip of the axon involves the same ordered recruitment of the core autophagic proteins as in non-polarised cells [139,140,141][12][13][14]. Nevertheless, some cell-type specificity occurs in neurons, with specialised proteins of the presynapse playing a role in the regulation of AV biogenesis. First, Bassoon binds to ATG5 and negatively regulates the ATG5-ATG12-ATG16 E3 ligase activity to suppress synaptic autophagy [140][13]. Thus, loss of Bassoon and Piccolo is associated with a local induction of autophagy, which contributes to the destruction of synaptic vesicles through the fusion of bouton-derived autophagosomes with somatic lysosomes. Secondly, the recruitment of ATG proteins at the synapse is calcium-dependent and mediated by phosphorylated Endophilin A (EndoA). Curved membranes induced by phosphorylated EndoA allows for the recruitment of ATG3, promoting LC3 lipidation and thus autophagosome formation [142,143][15][16]. To allow the local and constant biogenesis of autophagosomes, synaptojanin, a lipid phosphatase enriched in synaptic boutons, dephosphorylates PI3P to enable WIPI2 removal from nascent phagophores [144][17]. This recycling replenishes the WIPI2 soluble pool to form new phagophores.
In PD, EndoA phosphorylation has been described as deregulated due to mutations in the leucine-rich repeat serine/threonine-protein kinase 2 (LRRK2) [145][18]. LRRK2 mutation inhibits EndoA membrane association, endocytosis, and autophagy, leading to dopaminergic neuron degeneration [142,146][15][19]. The role of LRRK2 has been confirmed in flies expressing a phospho-deficient EndoA, which exhibit an inhibition of starvation-induced synaptic autophagy [142][15]. Moreover, EndoA mutation has also been identified as a PD risk variant and was shown to be mislocalised and to block the formation of autophagosomes at pre-synaptic terminals after stimulation and Ca2+ influx [147][20]. Another PD risk mutation in EndoA at the SH3 domain containing GRB2 like 2 (SH3GL2) has been described as disrupting the calcium sensing of SH3GL2. The resultant immobile protein cannot respond to calcium influx and therefore disrupts autophagy induction at the synapses [148][21]. A PD mutation in the SCA1 domain of synaptojanin inhibits its phosphatase activity. Mutant flies and iPSC-derived neurons from patients exhibit accumulation of WIPI proteins (Atg18 in flies) on nascent synaptic autophagosomes. This inhibits their completion, leading to their accumulation in the synapses [144][17].
1.1.2. Maturation during Transport
After their formation at the distal part of the axon, autophagosomes mature by fusing with acidic vesicles, such as early and late endosomes, to form amphisomes and with lysosomes to form autolysosomes (Figure 21B). The situation is complex, with different fusion events generating distinct AVs all along the axons. To summarise, autophagosomes formed in the growth cone first undergo bidirectional movement [149][22], which then switches to unidirectional movement along the axon. This change in motility depends on the maturation state of the autophagosome. The acquisition of endo-lysosomal markers such as Rab5, Rab7 and LAMP1, indicating a fusion with endosome or lysosome, is necessary for dynein-mediated retrograde transport in axons [149,150,151][22][23][24]. P62 has also been reported to be required for proper dynein motility and trafficking along microtubules (MTs) via a direct interaction with dynein [152][25]. Conversely, knock-down of p62, as well as pharmacological inhibition of dynein activity with ciliobrevin D, were found to reduce autolysosome formation [153][26]. Thus, the maturation process is coupled with axonal transport and generates a spatial gradient of organelle pH along the axon [154][27]. Most organelles in distal axons (86%) are neutral with a pH of >6.8, while the first 100 µm of the axon, corresponding to the proximal part, comprises 44% of neutral organelles and 56% acidic with a pH of <6.0. The dual-colour autophagic probe mCherry-GFP-LC3, which labels immature AVs in yellow and mature AVs in red due to GFP quenching in an acidic environment, has allowed for research into maturation and transport coupling [149][22]. This analysis revealed that immature AVs are more abundant in the distal part of the axon compared to the proximal part, which is mostly composed of mature AVs.
In the neuron, axonal transport of AVs, coupled with maturation, depends on MTs, the molecular motors dynein and kinesin, and adaptors. In a nutrient-deprived environment, the addition of depolymerising drugs such as nocodazole prevents degradative response by inhibiting the delivery of lysosomal hydrolytic enzymes to autophagosomes [155][28]. Furthermore, live imaging of autophagosome dynamics with GFP-LC3 confirms that MTs are required for their motility and fusion with lysosomes [156][29].
Autophagosomes, formed in the growth cone, initially undergo a bidirectional movement along MTs with the help of kinesin and dynein [149][22]. Little is known about autophagosome motility regulation in the distal tips prior to their exit. Disruption of the retrograde transport by impairing the recruitment of dynein adaptors can induce kinesin activation and bidirectional or anterograde transport [157,158,159][30][31][32]. Moreover, the LC3B phosphorylation state regulates the directional transport of AVs. Its phosphorylation by the serine/threonine-protein kinase 4 (STK4) promotes the recruitment of the FYVE and coiled-coil domain-containing protein 1 (FYCO1), a kinesin adaptor for anterograde transport [160,161][33][34].
The switch of mobility in the distal axon, from bidirectional/stationary to unidirectional retrograde transport is poorly understood. Studies suggest that fusion between autophagosomes and endosomes in the distal tip plays a role in dynein recruitment [151,162,163][24][35][36]. To allow for their exit from the distal tips through unidirectional retrograde transport, dynein is recruited to AVs through interaction with JNK-interacting protein (JIP) 1 (JIP1), a motor adaptor [157,164][30][37]. While JIP1 can bind either dynein or kinesin, its interaction with dynein and LC3 induces an inactivation of kinesin, therefore promoting retrograde transport from the distal to the middle part of the axon [157,165][30][38].
AV retrograde transport in the middle axon has been shown to depend on the Huntingtin-associated protein 1 (HAP1) and HTT [158][31]. HAP1 forms a motor complex with dynein and dynactin via conserved dynein- and dynactin-binding sites [159][32]. This dynein–dynactin–HAP1 complex interacts with AVs via the scaffolding protein HTT. Depleting HTT in neurons of dorsal root ganglion results in a significantly decreased percentage of retrograde AVs and a significant increase of bidirectional or stationary movements [158][31]. Loss of HAP1 also decreases the run speeds of retrograde-directed AVs. Likewise, abolishing HTT–HAP1 interaction disrupts autophagosome transport, resulting in more stationary AVs [158][31].
Finally, as the AVs move proximally, another motor protein adaptor, JIP3, initially known to be involved in endosome transport, regulates the retrograde transport of acidic AVs via dynein interaction [159,166,167,168,169][32][39][40][41][42]. JIP3 mutants generate an increased number of AVs in neurites in C. elegans, zebrafish, and cortical neurons due to defective retrograde transport [166,167,168,169][39][40][41][42]. These motor adaptors are recruited in an AV maturation-dependent manner. JIP1 and HAP1 have been found to essentially bind with immature AVs, while JIP3 associates with both mature and immature AVs [159][32], confirming the coupling of the transport and maturation of AVs in neurons. Mature AVs, mostly found in the proximal axon, were shown to exhibit more stationary/bidirectional or anterograde movements than immature AVs [149][22]. After entry into the soma, AVs seem to be unable to re-enter the axon, therefore promoting fusion with the degradative lysosomes that are enriched in the soma [130][1].
1.1.3. Degradation
The primary site of autophagosomal degradation is the soma, where lysosomes are concentrated (see part B). However, studies have revealed that lysosomes can also be present in the axons and fuse with autophagosomes outside the soma.
Lysosomes, labelled by LAMP1, have been observed in the axon and accumulate within the swollen axons of amyloid plaques in AD mice [168][41]. Often deficient in the main proteases (cathepsins B, D and L), these organelles have been argued to represent an early stage of lysosome maturation. They would result from the fusion of lysosomal precursors in the distal regions of axons, and, like autophagosomes, their maturation would require retrograde transport to the cell body. Indeed, luminal proteases are enriched in lysosomes present in the soma [187][43]. On the other hand, time-lapse imaging performed in microfluidic devices provides evidence for the presence of cathepsins D, B, and L in lysosomes within the axonal compartment. The anterograde transport of mature lysosomes formed in the soma, as seen by the movement of somal degradative lysosomes towards distal axons [188][44], is most likely sustaining the local degradation capacity at distal regions of the axon and at the synapse. Thus, disrupting the axonal delivery of degradative lysosomes through depletion of Arl8, a lysosomal kinesin adaptor, has been shown to induce axonal autophagic stress with accumulation of autophagosomes in distal axons [188][44]. The anterograde transport of mature lysosomes into the axon is facilitated by ER tubules, which promote kinesin-1-powered lysosome fission (as seen in autophagic lysosome reformation). ER tubule–lysosome contact has been reported to occur at the pre-axonal region, facilitating the entry of lysosomes in the axon. Disruption of these interactions causes an accumulation of enlarged and less motile lysosomes in the soma, without affecting their degradative capacities [189][45]. Altogether, these findings indicate that degradative lysosomes are dynamically delivered to distal axons during the development and maturation of neurons.
1.2. Autophagy in the Soma
Soma is the main site for degradation of autophagosome content, as degradative lysosomes are concentrated in the perinuclear area. As mentioned above, non-degradative lysosomes can be observed in all regions of neurons but competent lysosomes, identified by the activity of lysosomal hydrolases such as cathepsin D, are enriched in the soma. More precisely, 45% of LAMP1-labeled organelles in the soma contain cathepsin D. This number drops to 29% in dendrites and 30% in axons, suggesting that the percentage of degradative LAMP1 organelles is higher in the soma than in neuronal processes [190][46].
Interestingly, a population of large LC3-positive autophagosomes, exhibiting weak LAMP1 staining, has evidenced a novel biogenesis site for autophagosome in the soma [130][1]. Those structures were found to be stationary or oscillating within a small area of a few micrometres. Starvation does not increase the number of this autophagosome population. However, degradation inhibition with bafilomycin increases GFP-LC3 puncta in the soma but not in the axon. Very little is known regarding the molecular mechanisms and physiological roles of this specialised site of autophagosome biogenesis, but its existence is supported by studies performed on gigaxonin. This E3 ubiquitin ligase, mutated in giant axonal neuropathy (GAN) [191][47], was shown to control autophagosome elongation through ubiquitin-dependant degradation of ALTG16L1 [192][48]. In GAN neurons, the net production of autophagosomes is reduced as a result of ATG16L1 accumulation, whose location in the soma suggests a biogenesis site within this neuronal compartment [192][48].
Independent studies have shown that damaged or depolarised mitochondria within the axon are transported retrogradely towards the soma, where they are engulfed in newly formed autophagosomes and degraded [193,194][49][50].
1.3. Autophagy in Dendrites
Autophagy in dendrites has not been described as much as axonal autophagy.
Autophagosome biogenesis in dendrites is infrequent, with a weak AV density [120[40][51],167], indicating that autophagosomes observed in the soma mainly originate from the axon [139][12].
Autophagic activity in dendrites does not respond to starvation [130][1], but to neuronal activity. KCl-induced depolarisation induces an increase of the LC3-II/LC3-I ratio in cultured hippocampal neurons in a NMDAR- (N-methyl-D-aspartate receptor-) dependent manner and an increase of GFP-LC3 puncta in the soma and dendritic spines [195][52]. Activation of autophagosome biogenesis in dendrites has also been reported upon long-term depression with NMDA [196][53].
In contrast to the preferred retrograde transport in axons, autophagosomes in dendrites predominantly exhibit bidirectional transport [139][12]. In contrary to the axonal compartment, MTs within dendrites are equally oriented in each direction, which may explain the bidirectionality of AV transport [197][54]. KCl-induced depolarisation of neurons or pharmacological stimulation results in a very striking and rapid decrease of AV motility within dendrites by reducing their average speed [198][55]. Conversely, silencing synaptic activity with antagonists of excitatory AMPAR (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor) and NMDAR induces AV motility in dendrites. This effect of neuronal activity on AV motility is specific to dendrites. Live imaging reveals that stimulation of synaptic activity increases the percentage of AVs at the synapse, whereas its silencing decreases association of AVs to post-synaptic regions [198][55]. AV motility in dendrites dependents on dynein since treatment with Ciliobrevin-D, an inhibitor of dynein-mediated transport, increases the density of dendritic AV after synaptic stimulation [196][53].
Inhibition of autophagosome/lysosome fusion induces an increased autophagosome density in the dendritic shaft of pyramidal neurons, which is amplified after NMDA treatment. This observation confirms that autophagosomes are present in dendrites even under basal conditions and suggests that their clearance upon fusion with lysosomes limits their detection [195][52]. Indeed, dendritic autophagosomes are described as rapidly acquiring LAMP1, likely through fusion with late endosomes and lysosomes [198][55]. Furthermore, stimulation of synaptic activity increases the number of acidic organelles such as lysosome in dendrites [198,199][55][56]. This suggests that the increase of lysosomes after NMDA treatment may participate in the rapid dynamics of autophagic degradation [195][52]. As in the axon, transport and maturation of AVs in dendrites are interlinked, and inhibition of lysosome protease activity decreases their motility in dendrites [199][56].
The degradative abilities of acidic organelles have been confirmed by DQ-BSA (dye quenched-bovine serum albumin), an endocytosed cargo that fluoresces only upon proteolytic cleavage. While DQ-BSA–positive puncta are scarce in axons, they have been found to be present in dendrites and enriched in the soma [198][55]. This confirms the degradative ability of LAMP1-positive vesicles containing cathepsin-B observed in the soma and in the proximal portion of dendrites [200][57]. However, LAMP1-positive compartments further than 25 µm from the soma were rarely found to contain cathepsin-B. Those observations indicate that in the distal dendrites the degradative capacity is not sufficient for terminal degradation, which, similarly to the axon, occurs mostly in the soma [200][57].
2. Autophagy in Neurological Physiopathology
As demonstrated in the first two parts of this review, autophagy is an essential process for maintaining neuronal integrity. The initial evidence for the pivotal role of autophagy in nervous systems was found in mice, where neuronal depletion of ATG5 and ATG7 causes neurodegeneration with behavioural deficits, axonal swelling, and dendritic atrophy [93,94][58][59]. Moreover, deregulations in the autophagy pathway cause numerous NDDs, suggesting the role of this degradative pathway in neuronal development. In this section, the researchers review the current knowledge of the main roles of autophagy in neurological physiology, from stem cells homeostasis and neuronal development to synapse function and plasticity (Figure 32).
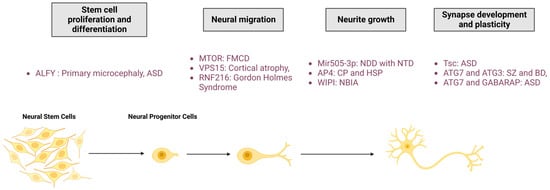
Figure 32. Roles of autophagy in neuronal physio(patho)logy. The autophagic process is fundamental for neuronal homeostasis and is implicated in stem cell proliferation and differentiation, neural migration, neurite outgrowth, and synapse development and plasticity. Mutation of proteins involved in autophagy pathway regulation leads to neurodevelopmental diseases. FMCD: Focal malformations of cortical development; NDD: Neurodevelopmental disorder; CP: Cerebral palsy; HSP: Hereditary spastic paraplegia; NBIA: Neurodegeneration with brain iron accumulation; ASD: Autism spectrum disorder; SZ: Schizophrenia; BD: Bipolar disorders; RNF216: Ring Finger Protein 216; Mir505-3p: miRNA 505-3p; and Tsc: tuberous sclerosis protein.
2.1. Neural Proliferation and Differentiation
Neurodevelopment in mammals is a complex process which starts in the neural stem cells (NSCs). They undergo symmetric cell division for self-renewal, proliferation to maintain stem cells niches, and asymmetric cell division to generate neural progenitor cells (NPCs). These progenitors are also able to self-renew and differentiate to produce neurons. Neurogenesis occurs during embryonic development and, to a lesser extent, in adulthood [201,202][60][61]. This process is regulated by numerous pathways, including autophagy [203][62].
Autophagy is necessary for maintaining NSC homeostasis and to control cell division. Indeed, autophagy has been shown to control the maintenance of NSCs in a quiescence state, since autophagy inhibition through ATG7 and ATG16L1 downregulation impairs differentiation and decreases neurogenesis [204][63]. These effects can be reversed through Beclin1 overexpression or under starvation. Epithelial v-like antigen 1 (Eva1a), a negative regulator of mTOR, is an important protein for NSC homeostasis, as Eva1a-depleted mice exhibit an inhibition of autophagy and an impairment of NSC self-renewal and differentiation in the cortex [205][64].
A premature transition between the symmetrical and asymmetrical division of NSCs leads to a decreased number of cortical neurons in the developing brain and causes microcephaly, a congenital NDD characterised by a reduced brain volume. This transition of cellular division is dependent on the autophagy adaptor protein ALFY [46][65]. Two different mouse models depleted for ALFY were shown to exhibit increased neural progenitor proliferation with increased cerebral size and a thinner neocortex in embryos [206,207][66][67]. Surprisingly, in humans, proliferation defects result in both microcephaly due to ALFY mutation [46][65] or macrocephaly due to WDFY3- (encoding for ALFY) haploinsufficiency, which has been associated with ASD [208,209][68][69] (Figure 32). Drosophila that express ALFY mutations found in patients affected by microcephaly have been described with reduced autophagy and premature asymmetric division leading to a reduced NCS pool [46][65].
The autophagy pathway plays a crucial role in NSC differentiation into NPCs and neurons. Protein levels of Beclin1, AMBRA1, ATG7 and LC3 have been found to progressively accumulate in the mouse embryo olfactory bulb during neuronal differentiation [210][70]. Moreover, inhibition of autophagy in mice through BECN1- (encoding for Beclin1) haploinsufficiency or AMBRA1 deficiency inhibits the differentiation of NSCs into NPCs, both in embryos and adults [210,211][70][71]. In adults, FIP200 has been found to be required for the maintenance and differentiation of postnatal NSCs. FIP200-depleted models show subventricular zone and dentate gyrus defects associated with impaired neurogenesis [212,213][72][73]. Autophagy inhibition by ATG5 downregulation increases the proliferation of cortical NPCs and inhibits their differentiation into neurons [214][74]. Additionally, the loss of ATG5 inhibits polarity acquisition of neural progenitors, which exhibit fewer and shorter neurites in the developing cortex.
2.2. Neural Migration
In non-neuronal and neuronal cells, motility requires assembly and disassembly of the focal adhesion. Several studies have evidenced the role of lysosomal degradation in focal adhesion recycling during migration. Autophagy-deficient cells have a reduced migration rate and increased focal adhesion number and size [215,216][75][76]. Moreover, the autophagic receptor NBR1 has been shown to promote focal adhesion disassembly and turnover through the autophagy pathway [215][75]. The essential role of autophagy in neuronal migration has been further confirmed in neuroblasts deficient for ATG5 or ATG12, which exhibit an accumulation of focal adhesion molecules such as Paxillin [217][77]. As a component of focal adhesion, the integrin steady-state is dictated by autophagy [218][78]. Indeed, VPS18 depletion leads to an upregulation of β1 integrin. Mice lacking VPS18 show neural migration defects in the cerebral cortex, hippocampus, and cerebellum [219][79]. As seen for ATG5 and ATG7 [93[58][59],94], VPS18 deficiency results in embryonic or early postnatal lethality and its neuronal-specific depletion leads to postnatal lethality and growth retardation [219][79], confirming the essential role of autophagic degradation during neuronal development.
During adult neurogenesis, autophagy has been found to be positively regulated by let-7 miRNA, permitting radial migration of newly formed olfactory bulb interneurons [220][80]. Let-7 downregulation prevents radial migration and maturation, which can be rescued by Beclin1 overexpression [220][80]. However, the RING finger E3 ligase RNF216 mediates Beclin1 ubiquitin-dependent degradation and promotes cell migration of immortalised gonadotropin-releasing hormone expressing neurons. RNF216 inhibition or autophagy activation inhibits neuronal migration [221][81]. It is nevertheless interesting to note that Beclin1 downregulation alone does not affect cell migration, suggesting another non-canonical role of Beclin1 during neurogenesis [221][81].
Several mutations of the autophagy pathway cause NDD in humans (Figure 32). MTOR mutations have been identified in patients affected by focal malformations of cortical development (FMCDs), a heterogeneous group of cortical abnormalities associated with epilepsy, intellectual disability, developmental delay, and autism [222,223,224,225,226,227][82][83][84][85][86][87]. MTOR mutations cause defective autophagy and disrupted migration of cortical neurons [228][88]. Patients carrying VPS15 mutations exhibit a wide range of symptoms, including cortical and optic nerve atrophy, cortical dysplasia, intellectual impairment, spasticity, ataxia, psychomotor delay, and late-onset epilepsy [31][89]. A VPS15 mutation has been shown to compromise the function of the PI3KC3 complex, leading to an accumulation of autophagic substrates [31][89]. RNF216 mutations have been identified in patients affected by Gordon Holmes syndrome, characterised by ataxia, dementia, and hypogonadotropic hypogonadism [229][90].
2.3. Neurite Outgrowth
The involvement of autophagy in neurite outgrowth has been evidenced for both axons and dendrites [230][91].
Autophagy can act as a negative regulator of axonal outgrowth. ATG12 is downregulated by the miRNA Mir505-3p, which inhibits autophagy, therefore promoting neural polarity and increasing axonal length and axonal branching [231][92]. Mir505-3p depletion increases the number of AVs and lowers the density of mitochondria, indicating increased mitophagy [231][92]. Axonal outgrowth, a high energy-demanding process, requires functional mitochondria throughout the axon, in order to provide a large amount of ATP [232,233][93][94]. Thus, it has been proposed that Mir505-3p locally downregulates axonal autophagy to maintain a sufficient mitochondria pool to promote axonal extension and branching [231][92]. This is consistent with previous studies reporting an excessive elongation of axons after autophagy inhibition through the depletion of ATG7 in cortical neurons [234][95] and of ATG2, ATG13, or ATG9 in C. elegans [137][10]. In addition, STX17 depletion has been shown to induce a reduction of axonal length in an AD mouse model [235][96].
Finally, several other studies reveal the opposing roles of autophagy on neurite outgrowth. Thus, ATG7 and/or ATG9 deficiency in mice does not increase axonal length but does impair nerve fibre formation with a larger contact site in axon terminals [236][97]. Moreover, pharmacological inhibition of autophagy decreases neurite length and branching complexity [237][98] of dorsal root ganglion neurons, and leads to a loss of neuronal polarity in hippocampal neurons, while its activation stimulates axonal growth [238][99]. During hypothalamus development, ATG7 depletion in neurons producing pro-opiomelanocortin induces alterations of neuronal projections that decrease their ability to extend axons during postnatal development [239][100].
The role of autophagy in dendrite outgrowth is more poorly studied than in axons. However, some studies have evidenced its crucial involvement in dendritic arborisation. AP-2, a key factor in ATG9 localisation [240][101] as for AP-4, has been described as an adaptor protein forming a complex between LC3 and dynein/dynactin, regulating autophagosome retrograde transport [241][102]. AP-2 depletion or downregulation leads to defective retrograde transport of AVs and impairs dendritic arborisation both in vitro and in vivo [241,242][102][103]. Interestingly, while AP-4 deficiency induces ATG9 mislocalisation in the axon, ATG9 in dendrites remains unaltered [135][8], confirming a compartment-dependent regulation of autophagy. The role of autophagy in dendrites is controversial. Downregulation of ATG5 and ATG7 was found to result in enlarged dendritic trees [243][104], while another study showed that basal autophagy is required for dendritic growth and branching [244][105]. Surprisingly, the latter study also showed that Atg1 (ULK1) overexpression decreases dendritic tree growth and terminal branching [244][105], indicating that both insufficient and excessive autophagy are deleterious for neuritic development.
Mutations and dysregulations of autophagy proteins may lead to neurodevelopmental and neurodegenerative diseases due to abnormalities in neurite outgrowth.
Neurodevelopmental abnormalities with neural tube closure defects, such as spina bifida, have been associated with the duplication of genomic segments containing Mir505-3p [245][106]. AP-4 mutations are causal for NDDs such as Cerebral Palsy and HSP [246,247][107][108], characterised by defects in axon outgrowth and neurodegeneration. Mimicking AP-4 loss-of-function in disease, AP-4 depletion was shown to reduce axonal ATG9A, leading to decreased autophagosomes in axons that may underly defective axonal extension [79,80,135][7][8][109]. Mutations of WIPI proteins cause NBIA, a NDD for which patients exhibit reduced white matter volume [248][110].
2.4. Synapse Development and Plasticity
Autophagy at synapses has a specific mode of regulation, as described in the previous section (Figure 21C). In this specific compartment, autophagy and neuronal activity are reciprocally regulated. In this section, the researchers will specifically discuss the roles of autophagy in regulating synaptic plasticity, from synaptogenesis to the recycling of neurotransmitter receptors (Figure 43).
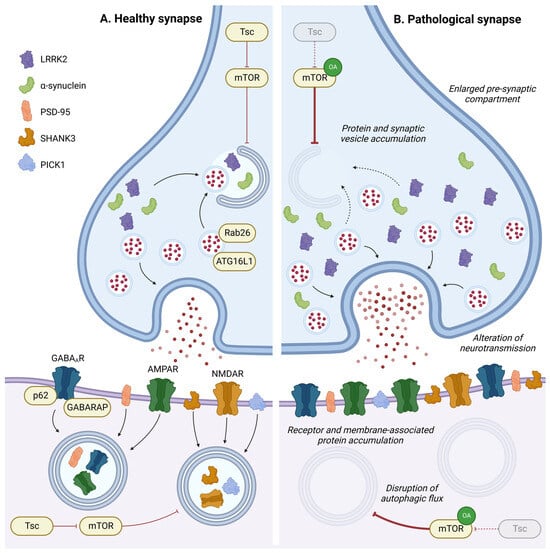
Figure 43. Role of autophagy in synapse homeostasis. In pre- or post-synaptic compartments, autophagy has a key role in regulating synaptic proteins or vesicles and is implicated in synaptic plasticity in response to environmental stimulation. Disruption in the autophagic process leads to accumulation of intracellular components, neurotransmitter receptors, and membrane-associated proteins in pre- or post-synaptic compartments, leading to an enlargement of the pre-synaptic compartment and alterations in neurotransmission. OA: Overactivation; GABAAR: gamma-aminobutyric acid receptor; AMPAR: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; and NMDAR: N-methyl-D-aspartate receptor.
Synaptic defects and malformations have been reported in vivo as a result of genetic autophagy impairments. In C. elegans, autophagy is required for clustering synaptic vesicle proteins and promoting presynaptic assembly in interneurons during development [137][10]. In the D. melanogaster learning centre (i.e., mushroom bodies), autophagy inhibition increases the size of presynaptic active zones [249][111]. Conversely, mutations in different ATG genes in D. melanogaster induce a reduction of neuromuscular junction (NMJ) size, while induction of autophagy by Atg1 (ULK1) overexpression leads to NMJ overgrowth [250][112]. Atg7−/− flies present an increase in ELKS-family protein Bruchpilot (BRP), an essential building block of the active zone scaffold, and an increase in active zone size leading to memory impairment [251][113]. In mice, motor neuron-specific deletion of ATG7 induces structural and functional defects within the NMJ [252][114]. By crossing ATG7-depleted mice with ALS mice, autophagy inhibition was shown to accelerate early neuromuscular denervation in pathological mice [252][114].
Altogether, these findings demonstrate that a finely regulated basal level of autophagy is required to structure synapses, both between neurons in the central nervous system and at the NMJ in the peripheral nervous system.
Autophagy deficiency in dopaminergic neurons by ATG7 depletion increases the size of dopaminergic synaptic terminals and induces striatal dopaminergic innervation, leading to increased dopaminergic neurotransmission [253][115]. This result indicates the involvement of autophagy in neurotransmission regulation at presynaptic sites. In agreement with this, stimulation of autophagy by rapamycin has been shown to decrease the number of synaptic vesicles through autophagic degradation in mice [253][115]. Moreover, ATG16L1 has been found to be recruited at the axonal terminal by the GTPase Rab26, which is enriched in synaptic vesicle clusters to mediate their autophagic degradation [254][116].
Several studies have pinpointed the roles of autophagy in the postsynaptic region. After neurogenesis, dendritic spines can be removed during postnatal development, in a process called spine pruning, which only stabilises specific synapses. Synapse morphology and dendritic spine remodelling rely on the autophagic degradation of key postsynaptic proteins such as SHANK3 (SH3 and multiple ankyrin repeat domains protein 3), PSD-95 (postsynaptic density protein 95), and PICK1 (protein interacting with C kinase 1) [255][117]. Autophagy activation in fragile X mice, which exhibit mTOR overactivation, allows for autophagy-dependent degradation of PSD-95 and corrects spine morphology and cognition deficits [256][118]. Overall, activation of autophagy signalling represents a strategy to normalise developmental dendritic spine pruning defects and social behaviours [257][119]. However, inhibition of autophagy via mTOR causes spine pruning defects. Thus, tuberous sclerosis protein (Tsc) haplo-insufficient mice studies, frequently used as an ASD model, result in a constitutive overactivation of mTOR and cause postnatal spine pruning alterations [257][119]. Additionally, Tsc1-deficient neurons exhibit alterations in excitatory functions characterised by an enhancement of postsynaptic glutamatergic functions due to an augmentation in the number of synapses [258][120]. Interestingly, Tsc1 deficiency in GABAergic interneurons of young adult mice results in a reduced number of synapses. This phenotype has been associated with a transient autophagy dysfunction in adolescent mice, presenting dysregulation in the expression of several autophagic markers (LC3-II, p62, ULK1) [259][121].
References
- Maday, S.; Holzbaur, E.L.F. Compartment-Specific Regulation of Autophagy in Primary Neurons. J. Neurosci. 2016, 36, 5933–5945.
- Bunge, M.B. Fine structure of nerve fibers and growth cones of isolated sympathetic neurons in culture. J. Cell Biol. 1973, 56, 713–735.
- Hollenbeck, P.J. Products of Endocytosis and Autophagy Are Retrieved from Axons by Regulated Retrograde Organelle Transport. J. Cell Biol. 1993, 121, 305–315.
- Maday, S.; Holzbaur, E.L.F. Autophagosome Assembly and Cargo Capture in the Distal Axon. Autophagy 2012, 8, 858–860.
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In Vivo Analysis of Autophagy in Response to Nutrient Starvation Using Transgenic Mice Expressing a Fluorescent Autophagosome Marker. Mol. Biol. Cell 2004, 15, 1101–1111.
- Matoba, K.; Kotani, T.; Tsutsumi, A.; Tsuji, T.; Mori, T.; Noshiro, D.; Sugita, Y.; Nomura, N.; Iwata, S.; Ohsumi, Y.; et al. Atg9 Is a Lipid Scramblase That Mediates Autophagosomal Membrane Expansion. Nat. Struct. Mol. Biol. 2020, 27, 1185–1193.
- De Pace, R.; Skirzewski, M.; Damme, M.; Mattera, R.; Mercurio, J.; Foster, A.M.; Cuitino, L.; Jarnik, M.; Hoffmann, V.; Morris, H.D.; et al. Altered Distribution of ATG9A and Accumulation of Axonal Aggregates in Neurons from a Mouse Model of AP-4 Deficiency Syndrome. PLoS Genet. 2018, 14, e1007363.
- Ivankovic, D.; Drew, J.; Lesept, F.; White, I.J.; López Doménech, G.; Tooze, S.A.; Kittler, J.T. Axonal Autophagosome Maturation Defect through Failure of ATG9A Sorting Underpins Pathology in AP-4 Deficiency Syndrome. Autophagy 2020, 16, 391–407.
- Yang, S.; Park, D.; Manning, L.; Hill, S.E.; Cao, M.; Xuan, Z.; Gonzalez, I.; Dong, Y.; Clark, B.; Shao, L.; et al. Presynaptic Autophagy Is Coupled to the Synaptic Vesicle Cycle via ATG-9. Neuron 2022, 110, 824–840.e10.
- Stavoe, A.K.H.; Hill, S.E.; Hall, D.H.; Colón-Ramos, D.A. KIF1A/UNC-104 Transports ATG-9 to Regulate Neurodevelopment and Autophagy at Synapses. Dev. Cell 2016, 38, 171–185.
- Xuan, Z.; Yang, S.; Clark, B.; Hill, S.E.; Manning, L.; Colón-Ramos, D.A. The Active Zone Protein Clarinet Regulates Synaptic Sorting of ATG-9 and Presynaptic Autophagy. PLoS Biol. 2023, 21, e3002030.
- Maday, S.; Holzbaur, E.L.F. Autophagosome Biogenesis in Primary Neurons Follows an Ordered and Spatially Regulated Pathway. Dev. Cell 2014, 30, 71–85.
- Okerlund, N.D.; Schneider, K.; Leal-Ortiz, S.; Montenegro-Venegas, C.; Kim, S.A.; Garner, L.C.; Waites, C.L.; Gundelfinger, E.D.; Reimer, R.J.; Garner, C.C. Bassoon Controls Presynaptic Autophagy through Atg5. Neuron 2017, 93, 897–913.e7.
- Stavoe, A.K.; Gopal, P.P.; Gubas, A.; Tooze, S.A.; Holzbaur, E.L. Expression of WIPI2B Counteracts Age-Related Decline in Autophagosome Biogenesis in Neurons. eLife 2019, 8, e44219.
- Soukup, S.-F.; Kuenen, S.; Vanhauwaert, R.; Manetsberger, J.; Hernández-Díaz, S.; Swerts, J.; Schoovaerts, N.; Vilain, S.; Gounko, N.V.; Vints, K.; et al. A LRRK2-Dependent EndophilinA Phosphoswitch Is Critical for Macroautophagy at Presynaptic Terminals. Neuron 2016, 92, 829–844.
- Murdoch, J.D.; Rostosky, C.M.; Gowrisankaran, S.; Arora, A.S.; Soukup, S.-F.; Vidal, R.; Capece, V.; Freytag, S.; Fischer, A.; Verstreken, P.; et al. Endophilin-A Deficiency Induces the Foxo3a-Fbxo32 Network in the Brain and Causes Dysregulation of Autophagy and the Ubiquitin-Proteasome System. Cell Rep. 2016, 17, 1071–1086.
- Vanhauwaert, R.; Kuenen, S.; Masius, R.; Bademosi, A.; Manetsberger, J.; Schoovaerts, N.; Bounti, L.; Gontcharenko, S.; Swerts, J.; Vilain, S.; et al. The SAC1 Domain in Synaptojanin Is Required for Autophagosome Maturation at Presynaptic Terminals. EMBO J. 2017, 36, 1392–1411.
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607.
- Matta, S.; Van Kolen, K.; da Cunha, R.; van den Bogaart, G.; Mandemakers, W.; Miskiewicz, K.; De Bock, P.-J.; Morais, V.A.; Vilain, S.; Haddad, D.; et al. LRRK2 Controls an EndoA Phosphorylation Cycle in Synaptic Endocytosis. Neuron 2012, 75, 1008–1021.
- Bademosi, A.T.; Decet, M.; Kuenen, S.; Calatayud, C.; Swerts, J.; Gallego, S.F.; Schoovaerts, N.; Karamanou, S.; Louros, N.; Martin, E.; et al. EndophilinA-Dependent Coupling between Activity-Induced Calcium Influx and Synaptic Autophagy Is Disrupted by a Parkinson-Risk Mutation. Neuron 2023, 111, 1402–1422.e13.
- Decet, M.; Soukup, S.-F. Endophilin-A/SH3GL2 Calcium Switch for Synaptic Autophagy Induction Is Impaired by a Parkinson’s Risk Variant. Autophagy, 2023; 1–3, online ahead of print.
- Maday, S.; Wallace, K.E.; Holzbaur, E.L.F. Autophagosomes Initiate Distally and Mature during Transport toward the Cell Soma in Primary Neurons. J. Cell Biol. 2012, 196, 407–417.
- Lee, S.; Sato, Y.; Nixon, R.A. Lysosomal Proteolysis Inhibition Selectively Disrupts Axonal Transport of Degradative Organelles and Causes an Alzheimer’s-Like Axonal Dystrophy. J. Neurosci. 2011, 31, 7817–7830.
- Cheng, X.-T.; Zhou, B.; Lin, M.-Y.; Cai, Q.; Sheng, Z.-H. Axonal Autophagosomes Recruit Dynein for Retrograde Transport through Fusion with Late Endosomes. J. Cell Biol. 2015, 209, 377–386.
- Calderilla-Barbosa, L.; Seibenhener, M.L.; Du, Y.; Diaz-Meco, M.-T.; Moscat, J.; Yan, J.; Wooten, M.W.; Wooten, M.C. Interaction of SQSTM1 with the Motor Protein Dynein: SQSTM1 Is Required for Normal Dynein Function and Trafficking. J. Cell Sci. 2014, 127, 4052–4063.
- Zhang, L.; Fang, Y.; Cheng, X.; Lian, Y.; Xu, H. Interaction between TRPML1 and P62 in Regulating Autophagosome-Lysosome Fusion and Impeding Neuroaxonal Dystrophy in Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2022, 2022, 8096009.
- Overly, C.C.; Lee, K.D.; Berthiaume, E.; Hollenbeck, P.J. Quantitative Measurement of Intraorganelle pH in the Endosomal-Lysosomal Pathway in Neurons by Using Ratiometric Imaging with Pyranine. Proc. Natl. Acad. Sci. USA 1995, 92, 3156–3160.
- Aplin, A.; Jasionowski, T.; Tuttle, D.L.; Lenk, S.E.; Dunn, W.A. Cytoskeletal Elements Are Required for the Formation and Maturation of Autophagic Vacuoles. J. Cell. Physiol. 1992, 152, 458–466.
- Köchl, R.; Hu, X.W.; Chan, E.Y.W.; Tooze, S.A. Microtubules Facilitate Autophagosome Formation and Fusion of Autophagosomes with Endosomes. Traffic 2006, 7, 129–145.
- Fu, M.; Nirschl, J.J.; Holzbaur, E.L.F. LC3 Binding to the Scaffolding Protein JIP1 Regulates Processive Dynein-Driven Transport of Autophagosomes. Dev. Cell 2014, 29, 577–590.
- Wong, Y.C.; Holzbaur, E.L.F. The Regulation of Autophagosome Dynamics by Huntingtin and HAP1 Is Disrupted by Expression of Mutant Huntingtin, Leading to Defective Cargo Degradation. J. Neurosci. 2014, 34, 1293–1305.
- Cason, S.E.; Carman, P.J.; Van Duyne, C.; Goldsmith, J.; Dominguez, R.; Holzbaur, E.L.F. Sequential Dynein Effectors Regulate Axonal Autophagosome Motility in a Maturation-Dependent Pathway. J. Cell Biol. 2021, 220, e202010179.
- Nieto-Torres, J.L.; Shanahan, S.-L.; Chassefeyre, R.; Chaiamarit, T.; Zaretski, S.; Landeras-Bueno, S.; Verhelle, A.; Encalada, S.E.; Hansen, M. LC3B Phosphorylation Regulates FYCO1 Binding and Directional Transport of Autophagosomes. Curr. Biol. CB 2021, 31, 3440–3449.e7.
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.-A.; Lamark, T.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. FYCO1 Is a Rab7 Effector That Binds to LC3 and PI3P to Mediate Microtubule plus End–Directed Vesicle Transport. J. Cell Biol. 2010, 188, 253–269.
- Jordens, I.; Fernandez-Borja, M.; Marsman, M.; Dusseljee, S.; Janssen, L.; Calafat, J.; Janssen, H.; Wubbolts, R.; Neefjes, J. The Rab7 Effector Protein RILP Controls Lysosomal Transport by Inducing the Recruitment of Dynein-Dynactin Motors. Curr. Biol. 2001, 11, 1680–1685.
- Wijdeven, R.H.; Janssen, H.; Nahidiazar, L.; Janssen, L.; Jalink, K.; Berlin, I.; Neefjes, J. Cholesterol and ORP1L-Mediated ER Contact Sites Control Autophagosome Transport and Fusion with the Endocytic Pathway. Nat. Commun. 2016, 7, 11808.
- Fu, M.; Holzbaur, E.L.F. JIP1 Regulates the Directionality of APP Axonal Transport by Coordinating Kinesin and Dynein Motors. J. Cell Biol. 2013, 202, 495–508.
- Fu, M.; Holzbaur, E.L.F. MAPK8IP1/JIP1 Regulates the Trafficking of Autophagosomes in Neurons. Autophagy 2014, 10, 2079–2081.
- Brown, H.M.; Van Epps, H.A.; Goncharov, A.; Grant, B.D.; Jin, Y. The JIP3 Scaffold Protein UNC-16 Regulates RAB-5 Dependent Membrane Trafficking at C. elegans Synapses. Dev. Neurobiol. 2009, 69, 174–190.
- Drerup, C.M.; Nechiporuk, A.V. JNK-Interacting Protein 3 Mediates the Retrograde Transport of Activated c-Jun N-Terminal Kinase and Lysosomes. PLoS Genet. 2013, 9, e1003303.
- Gowrishankar, S.; Wu, Y.; Ferguson, S.M. Impaired JIP3-Dependent Axonal Lysosome Transport Promotes Amyloid Plaque Pathology. J. Cell Biol. 2017, 216, 3291–3305.
- Hill, S.E.; Kauffman, K.J.; Krout, M.; Richmond, J.E.; Melia, T.J.; Colón-Ramos, D.A. Maturation and Clearance of Autophagosomes in Neurons Depends on a Specific Cysteine Protease Isoform, ATG-4.2. Dev. Cell 2019, 49, 251–266.e8.
- Gowrishankar, S.; Yuan, P.; Wu, Y.; Schrag, M.; Paradise, S.; Grutzendler, J.; De Camilli, P.; Ferguson, S.M. Massive Accumulation of Luminal Protease-Deficient Axonal Lysosomes at Alzheimer’s Disease Amyloid Plaques. Proc. Natl. Acad. Sci. USA 2015, 112, E3699–E3708.
- Farfel-Becker, T.; Roney, J.C.; Cheng, X.-T.; Li, S.; Cuddy, S.R.; Sheng, Z.-H. Neuronal Soma-Derived Degradative Lysosomes Are Continuously Delivered to Distal Axons to Maintain Local Degradation Capacity. Cell Rep. 2019, 28, 51–64.e4.
- Özkan, N.; Koppers, M.; van Soest, I.; van Harten, A.; Jurriens, D.; Liv, N.; Klumperman, J.; Kapitein, L.C.; Hoogenraad, C.C.; Farías, G.G. ER—Lysosome Contacts at a Pre-Axonal Region Regulate Axonal Lysosome Availability. Nat. Commun. 2021, 12, 4493.
- Cheng, X.-T.; Xie, Y.-X.; Zhou, B.; Huang, N.; Farfel-Becker, T.; Sheng, Z.-H. Characterization of LAMP1-Labeled Nondegradative Lysosomal and Endocytic Compartments in Neurons. J. Cell Biol. 2018, 217, 3127–3139.
- Bomont, P.; Cavalier, L.; Blondeau, F.; Hamida, C.B.; Belal, S.; Tazir, M.; Demir, E.; Topaloglu, H.; Korinthenberg, R.; Tüysüz, B.; et al. The Gene Encoding Gigaxonin, a New Member of the Cytoskeletal BTB/Kelch Repeat Family, Is Mutated in Giant Axonal Neuropathy. Nat. Genet. 2000, 26, 370–374.
- Scrivo, A.; Codogno, P.; Bomont, P. Gigaxonin E3 Ligase Governs ATG16L1 Turnover to Control Autophagosome Production. Nat. Commun. 2019, 10, 780.
- Cai, Q.; Zakaria, H.M.; Simone, A.; Sheng, Z.-H. Spatial Parkin Translocation and Degradation of Damaged Mitochondria via Mitophagy in Live Cortical Neurons. Curr. Biol. 2012, 22, 545–552.
- Zheng, Y.; Zhang, X.; Wu, X.; Jiang, L.; Ahsan, A.; Ma, S.; Xiao, Z.; Han, F.; Qin, Z.-H.; Hu, W.; et al. Somatic Autophagy of Axonal Mitochondria in Ischemic Neurons. J. Cell Biol. 2019, 218, 1891–1907.
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The Hairpin-Type Tail-Anchored SNARE Syntaxin 17 Targets to Autophagosomes for Fusion with Endosomes/Lysosomes. Cell 2012, 151, 1256–1269.
- Shehata, M.; Matsumura, H.; Okubo-Suzuki, R.; Ohkawa, N.; Inokuchi, K. Neuronal Stimulation Induces Autophagy in Hippocampal Neurons That Is Involved in AMPA Receptor Degradation after Chemical Long-Term Depression. J. Neurosci. 2012, 32, 10413–10422.
- Kallergi, E.; Daskalaki, A.-D.; Kolaxi, A.; Camus, C.; Ioannou, E.; Mercaldo, V.; Haberkant, P.; Stein, F.; Sidiropoulou, K.; Dalezios, Y.; et al. Dendritic Autophagy Degrades Postsynaptic Proteins and Is Required for Long-Term Synaptic Depression in Mice. Nat. Commun. 2022, 13, 680.
- Baas, P.W.; Black, M.M.; Banker, G.A. Changes in Microtubule Polarity Orientation during the Development of Hippocampal Neurons in Culture. J. Cell Biol. 1989, 109, 3085–3094.
- Kulkarni, V.V.; Anand, A.; Herr, J.B.; Miranda, C.; Vogel, M.C.; Maday, S. Synaptic Activity Controls Autophagic Vacuole Motility and Function in Dendrites. J. Cell Biol. 2021, 220, e202002084.
- Goo, M.S.; Sancho, L.; Slepak, N.; Boassa, D.; Deerinck, T.J.; Ellisman, M.H.; Bloodgood, B.L.; Patrick, G.N. Activity-Dependent Trafficking of Lysosomes in Dendrites and Dendritic Spines. J. Cell Biol. 2017, 216, 2499–2513.
- Yap, C.C.; Digilio, L.; McMahon, L.P.; Garcia, A.D.R.; Winckler, B. Degradation of Dendritic Cargos Requires Rab7-Dependent Transport to Somatic Lysosomes. J. Cell Biol. 2018, 217, 3141–3159.
- Komatsu, M.; Wang, Q.J.; Holstein, G.R.; Friedrich, V.L.; Iwata, J.; Kominami, E.; Chait, B.T.; Tanaka, K.; Yue, Z. Essential Role for Autophagy Protein Atg7 in the Maintenance of Axonal Homeostasis and the Prevention of Axonal Degeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 14489–14494.
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of Basal Autophagy in Neural Cells Causes Neurodegenerative Disease in Mice. Nature 2006, 441, 885–889.
- Giehl, K.M. Neuronal Development. Prog. Exp. Tumor Res. 2007, 39, 1–29.
- Molnár, Z.; Clowry, G.J.; Šestan, N.; Alzu’bi, A.; Bakken, T.; Hevner, R.F.; Hüppi, P.S.; Kostović, I.; Rakic, P.; Anton, E.S.; et al. New Insights into the Development of the Human Cerebral Cortex. J. Anat. 2019, 235, 432–451.
- Fleming, A.; Rubinsztein, D.C. Autophagy in Neuronal Development and Plasticity. Trends Neurosci. 2020, 43, 767–779.
- Wu, X.; Fleming, A.; Ricketts, T.; Pavel, M.; Virgin, H.; Menzies, F.M.; Rubinsztein, D.C. Autophagy Regulates Notch Degradation and Modulates Stem Cell Development and Neurogenesis. Nat. Commun. 2016, 7, 10533.
- Li, M.; Lu, G.; Hu, J.; Shen, X.; Ju, J.; Gao, Y.; Qu, L.; Xia, Y.; Chen, Y.; Bai, Y. EVA1A/TMEM166 Regulates Embryonic Neurogenesis by Autophagy. Stem Cell Rep. 2016, 6, 396–410.
- Kadir, R.; Harel, T.; Markus, B.; Perez, Y.; Bakhrat, A.; Cohen, I.; Volodarsky, M.; Feintsein-Linial, M.; Chervinski, E.; Zlotogora, J.; et al. ALFY-Controlled DVL3 Autophagy Regulates Wnt Signaling, Determining Human Brain Size. PLoS Genet. 2016, 12, e1005919.
- Le Duc, D.; Giulivi, C.; Hiatt, S.M.; Napoli, E.; Panoutsopoulos, A.; Harlan De Crescenzo, A.; Kotzaeridou, U.; Syrbe, S.; Anagnostou, E.; Azage, M.; et al. Pathogenic WDFY3 Variants Cause Neurodevelopmental Disorders and Opposing Effects on Brain Size. Brain 2019, 142, 2617–2630.
- Orosco, L.A.; Ross, A.P.; Cates, S.L.; Scott, S.E.; Wu, D.; Sohn, J.; Pleasure, D.; Pleasure, S.J.; Adamopoulos, I.E.; Zarbalis, K.S. Loss of Wdfy3 in Mice Alters Cerebral Cortical Neurogenesis Reflecting Aspects of the Autism Pathology. Nat. Commun. 2014, 5, 4692.
- Jacquemont, M.; Sanlaville, D.; Redon, R.; Raoul, O.; Cormier-Daire, V.; Lyonnet, S.; Amiel, J.; Merrer, M.L.; Heron, D.; de Blois, M.; et al. Array-based Comparative Genomic Hybridisation Identifies High Frequency of Cryptic Chromosomal Rearrangements in Patients with Syndromic Autism Spectrum Disorders. J. Med. Genet. 2006, 43, 843–849.
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.; Narzisi, G.; Leotta, A.; et al. De Novo Gene Disruptions in Children on the Autistic Spectrum. Neuron 2012, 74, 285–299.
- Vázquez, P.; Arroba, A.I.; Cecconi, F.; De La Rosa, E.J.; Boya, P.; De Pablo, F. Atg5 and Ambra1 Differentially Modulate Neurogenesis in Neural Stem Cells. Autophagy 2012, 8, 187–199.
- Yazdankhah, M.; Farioli-Vecchioli, S.; Tonchev, A.B.; Stoykova, A.; Cecconi, F. The Autophagy Regulators Ambra1 and Beclin 1 Are Required for Adult Neurogenesis in the Brain Subventricular Zone. Cell Death Dis. 2014, 5, e1403.
- Wang, C.; Liang, C.-C.; Bian, Z.C.; Zhu, Y.; Guan, J.-L. FIP200 Is Required for Maintenance and Differentiation of Postnatal Neural Stem Cells. Nat. Neurosci. 2013, 16, 532–542.
- Wang, C.; Yeo, S.; Haas, M.A.; Guan, J.-L. Autophagy Gene FIP200 in Neural Progenitors Non-Cell Autonomously Controls Differentiation by Regulating Microglia. J. Cell Biol. 2017, 216, 2581–2596.
- Lv, X.; Jiang, H.; Li, B.; Liang, Q.; Wang, S.; Zhao, Q.; Jiao, J. The Crucial Role of Atg5 in Cortical Neurogenesis During Early Brain Development. Sci. Rep. 2014, 4, 6010.
- Kenific, C.M.; Stehbens, S.J.; Goldsmith, J.; Leidal, A.M.; Faure, N.; Ye, J.; Wittmann, T.; Debnath, J. NBR1 Enables Autophagy-Dependent Focal Adhesion Turnover. J. Cell Biol. 2016, 212, 577–590.
- Sharifi, M.N.; Mowers, E.E.; Drake, L.E.; Collier, C.; Chen, H.; Zamora, M.; Mui, S.; Macleod, K.F. Autophagy Promotes Focal Adhesion Disassembly and Cell Motility of Metastatic Tumor Cells through the Direct Interaction of Paxillin with LC3. Cell Rep. 2016, 15, 1660–1672.
- Bressan, C.; Pecora, A.; Gagnon, D.; Snapyan, M.; Labrecque, S.; De Koninck, P.; Parent, M.; Saghatelyan, A. The Dynamic Interplay between ATP/ADP Levels and Autophagy Sustain Neuronal Migration in Vivo. eLife 2020, 9, e56006.
- Molnár, M.; Sőth, Á.; Simon-Vecsei, Z. Pathways of Integrins in the Endo-Lysosomal System. Biol. Futura 2022, 73, 171–185.
- Peng, C.; Ye, J.; Yan, S.; Kong, S.; Shen, Y.; Li, C.; Li, Q.; Zheng, Y.; Deng, K.; Xu, T.; et al. Ablation of Vacuole Protein Sorting 18 (Vps18) Gene Leads to Neurodegeneration and Impaired Neuronal Migration by Disrupting Multiple Vesicle Transport Pathways to Lysosomes. J. Biol. Chem. 2012, 287, 32861–32873.
- Petri, R.; Pircs, K.; Jönsson, M.E.; Åkerblom, M.; Brattås, P.L.; Klussendorf, T.; Jakobsson, J. Let-7 Regulates Radial Migration of New-born Neurons through Positive Regulation of Autophagy. EMBO J. 2017, 36, 1379–1391.
- Li, F.; Li, D.; Liu, H.; Cao, B.-B.; Jiang, F.; Chen, D.-N.; Li, J.-D. RNF216 Regulates the Migration of Immortalized GnRH Neurons by Suppressing Beclin1-Mediated Autophagy. Front. Endocrinol. 2019, 10, 12.
- Baybis, M.; Yu, J.; Lee, A.; Golden, J.A.; Weiner, H.; McKhann, G.; Aronica, E.; Crino, P.B. mTOR Cascade Activation Distinguishes Tubers from Focal Cortical Dysplasia. Ann. Neurol. 2004, 56, 478–487.
- Ljungberg, M.C.; Bhattacharjee, M.B.; Lu, Y.; Armstrong, D.L.; Yoshor, D.; Swann, J.W.; Sheldon, M.; D’Arcangelo, G. Activation of Mammalian Target of Rapamycin in Cytomegalic Neurons of Human Cortical Dysplasia. Ann. Neurol. 2006, 60, 420–429.
- Lee, J.H.; Huynh, M.; Silhavy, J.L.; Kim, S.; Dixon-Salazar, T.; Heiberg, A.; Scott, E.; Bafna, V.; Hill, K.J.; Collazo, A.; et al. De Novo Somatic Mutations in Components of the PI3K-AKT3-mTOR Pathway Cause Hemimegalencephaly. Nat. Genet. 2012, 44, 941–945.
- Leventer, R.J.; Scerri, T.; Marsh, A.P.L.; Pope, K.; Gillies, G.; Maixner, W.; MacGregor, D.; Harvey, A.S.; Delatycki, M.B.; Amor, D.J.; et al. Hemispheric Cortical Dysplasia Secondary to a Mosaic Somatic Mutation in MTOR. Neurology 2015, 84, 2029–2032.
- Lim, J.S.; Kim, W.; Kang, H.-C.; Kim, S.H.; Park, A.H.; Park, E.K.; Cho, Y.-W.; Kim, S.; Kim, H.M.; Kim, J.A.; et al. Brain Somatic Mutations in MTOR Cause Focal Cortical Dysplasia Type II Leading to Intractable Epilepsy. Nat. Med. 2015, 21, 395–400.
- Nakashima, M.; Saitsu, H.; Takei, N.; Tohyama, J.; Kato, M.; Kitaura, H.; Shiina, M.; Shirozu, H.; Masuda, H.; Watanabe, K.; et al. Somatic Mutations in the MTOR Gene Cause Focal Cortical Dysplasia Type IIb. Ann. Neurol. 2015, 78, 375–386.
- Park, S.M.; Lim, J.S.; Ramakrishina, S.; Kim, S.H.; Kim, W.K.; Lee, J.; Kang, H.-C.; Reiter, J.F.; Kim, D.S.; Kim, H.; et al. Brain Somatic Mutations in MTOR Disrupt Neuronal Ciliogenesis, Leading to Focal Cortical Dyslamination. Neuron 2018, 99, 83–97.e7.
- Gstrein, T.; Edwards, A.; Přistoupilová, A.; Leca, I.; Breuss, M.; Pilat-Carotta, S.; Hansen, A.H.; Tripathy, R.; Traunbauer, A.K.; Hochstoeger, T.; et al. Mutations in Vps15 Perturb Neuronal Migration in Mice and Are Associated with Neurodevelopmental Disease in Humans. Nat. Neurosci. 2018, 21, 207–217.
- Margolin, D.H.; Kousi, M.; Chan, Y.-M.; Lim, E.T.; Schmahmann, J.D.; Hadjivassiliou, M.; Hall, J.E.; Adam, I.; Dwyer, A.; Plummer, L.; et al. Ataxia, Dementia, and Hypogonadotropism Caused by Disordered Ubiquitination. N. Engl. J. Med. 2013, 368, 1992–2003.
- Garza-Lombó, C.; Gonsebatt, M.E. Mammalian Target of Rapamycin: Its Role in Early Neural Development and in Adult and Aged Brain Function. Front. Cell. Neurosci. 2016, 10, 157.
- Yang, K.; Yu, B.; Cheng, C.; Cheng, T.; Yuan, B.; Li, K.; Xiao, J.; Qiu, Z.; Zhou, Y. Mir505–3p Regulates Axonal Development via Inhibiting the Autophagy Pathway by Targeting Atg12. Autophagy 2017, 13, 1679–1696.
- Courchet, J.; Lewis, T.L.; Lee, S.; Courchet, V.; Liou, D.-Y.; Aizawa, S.; Polleux, F. Terminal Axon Branching Is Regulated by the LKB1-NUAK1 Kinase Pathway via Presynaptic Mitochondrial Capture. Cell 2013, 153, 1510–1525.
- Spillane, M.; Ketschek, A.; Merianda, T.T.; Twiss, J.L.; Gallo, G. Mitochondria Coordinate Sites of Axon Branching through Localized Intra-Axonal Protein Synthesis. Cell Rep. 2013, 5, 1564–1575.
- Ban, B.-K.; Jun, M.-H.; Ryu, H.-H.; Jang, D.-J.; Ahmad, S.T.; Lee, J.-A. Autophagy Negatively Regulates Early Axon Growth in Cortical Neurons. Mol. Cell. Biol. 2013, 33, 3907–3919.
- Zhang, L.; Fang, Y.; Zhao, X.; Zheng, Y.; Ma, Y.; Li, S.; Huang, Z.; Li, L. BRUCE Silencing Leads to Axonal Dystrophy by Repressing Autophagosome-Lysosome Fusion in Alzheimer’s Disease. Transl. Psychiatry 2021, 11, 421.
- Yamaguchi, J.; Suzuki, C.; Nanao, T.; Kakuta, S.; Ozawa, K.; Tanida, I.; Saitoh, T.; Sunabori, T.; Komatsu, M.; Tanaka, K.; et al. Atg9a Deficiency Causes Axon-Specific Lesions Including Neuronal Circuit Dysgenesis. Autophagy 2018, 14, 764–777.
- Clarke, J.; Mearow, K. Autophagy Inhibition in Endogenous and Nutrient-deprived Conditions Reduces Dorsal Root Ganglia Neuron Survival and Neurite Growth in Vitro. J. Neurosci. Res. 2016, 94, 653–670.
- Wojnacki, J.; Nola, S.; Bun, P.; Cholley, B.; Filippini, F.; Pressé, M.T.; Lipecka, J.; Man Lam, S.; N’guyen, J.; Simon, A.; et al. Role of VAMP7-Dependent Secretion of Reticulon 3 in Neurite Growth. Cell Rep. 2020, 33, 108536.
- Coupé, B.; Ishii, Y.; Dietrich, M.O.; Komatsu, M.; Horvath, T.L.; Bouret, S.G. Loss of Autophagy in Pro-Opiomelanocortin Neurons Perturbs Axon Growth and Causes Metabolic Dysregulation. Cell Metab. 2012, 15, 247–255.
- Popovic, D.; Dikic, I. TBC1D5 and the AP2 Complex Regulate ATG9 Trafficking and Initiation of Autophagy. EMBO Rep. 2014, 15, 392–401.
- Kononenko, N.L.; Claßen, G.A.; Kuijpers, M.; Puchkov, D.; Maritzen, T.; Tempes, A.; Malik, A.R.; Skalecka, A.; Bera, S.; Jaworski, J.; et al. Retrograde Transport of TrkB-Containing Autophagosomes via the Adaptor AP-2 Mediates Neuronal Complexity and Prevents Neurodegeneration. Nat. Commun. 2017, 8, 14819.
- Koscielny, A.; Malik, A.R.; Liszewska, E.; Zmorzynska, J.; Tempes, A.; Tarkowski, B.; Jaworski, J. Adaptor Complex 2 Controls Dendrite Morphology via mTOR-Dependent Expression of GluA2. Mol. Neurobiol. 2018, 55, 1590–1606.
- Kijak, E.; Pyza, E. TOR Signaling Pathway and Autophagy Are Involved in the Regulation of Circadian Rhythms in Behavior and Plasticity of L2 Interneurons in the Brain of Drosophila Melanogaster. PLoS ONE 2017, 12, e0171848.
- Clark, S.G.; Graybeal, L.L.; Bhattacharjee, S.; Thomas, C.; Bhattacharya, S.; Cox, D.N. Basal Autophagy Is Required for Promoting Dendritic Terminal Branching in Drosophila Sensory Neurons. PLoS ONE 2018, 13, e0206743.
- Hol, F.A.; Schepens, M.T.; van Beersum, S.E.; Redolfi, E.; Affer, M.; Vezzoni, P.; Hamel, B.C.; Karnes, P.S.; Mariman, E.C.; Zucchi, I. Identification and Characterization of an Xq26-Q27 Duplication in a Family with Spina Bifida and Panhypopituitarism Suggests the Involvement of Two Distinct Genes. Genomics 2000, 69, 174–181.
- Abou Jamra, R.; Philippe, O.; Raas-Rothschild, A.; Eck, S.H.; Graf, E.; Buchert, R.; Borck, G.; Ekici, A.; Brockschmidt, F.F.; Nöthen, M.M.; et al. Adaptor Protein Complex 4 Deficiency Causes Severe Autosomal-Recessive Intellectual Disability, Progressive Spastic Paraplegia, Shy Character, and Short Stature. Am. J. Hum. Genet. 2011, 88, 788–795.
- Moreno-De-Luca, A.; Helmers, S.L.; Mao, H.; Burns, T.G.; Melton, A.M.A.; Schmidt, K.R.; Fernhoff, P.M.; Ledbetter, D.H.; Martin, C.L. Adaptor Protein Complex-4 (AP-4) Deficiency Causes a Novel Autosomal Recessive Cerebral Palsy Syndrome with Microcephaly and Intellectual Disability. J. Med. Genet. 2011, 48, 141–144.
- Mattera, R.; Park, S.Y.; De Pace, R.; Guardia, C.M.; Bonifacino, J.S. AP-4 Mediates Export of ATG9A from the Trans -Golgi Network to Promote Autophagosome Formation. Proc. Natl. Acad. Sci. USA 2017, 114, E10697–E10706.
- Suleiman, J.; Allingham-Hawkins, D.; Hashem, M.; Shamseldin, H.E.; Alkuraya, F.S.; El-Hattab, A.W. WDR45B -Related Intellectual Disability, Spastic Quadriplegia, Epilepsy, and Cerebral Hypoplasia: A Consistent Neurodevelopmental Syndrome: SULEIMAN et Al. Clin. Genet. 2018, 93, 360–364.
- Bhukel, A.; Beuschel, C.B.; Maglione, M.; Lehmann, M.; Juhász, G.; Madeo, F.; Sigrist, S.J. Autophagy within the Mushroom Body Protects from Synapse Aging in a Non-Cell Autonomous Manner. Nat. Commun. 2019, 10, 1318.
- Shen, W.; Ganetzky, B. Autophagy Promotes Synapse Development in Drosophila. J. Cell Biol. 2009, 187, 71–79.
- Gupta, V.K.; Pech, U.; Bhukel, A.; Fulterer, A.; Ender, A.; Mauermann, S.F.; Andlauer, T.F.M.; Antwi-Adjei, E.; Beuschel, C.; Thriene, K.; et al. Spermidine Suppresses Age-Associated Memory Impairment by Preventing Adverse Increase of Presynaptic Active Zone Size and Release. PLoS Biol. 2016, 14, e1002563.
- Rudnick, N.D.; Griffey, C.J.; Guarnieri, P.; Gerbino, V.; Wang, X.; Piersaint, J.A.; Tapia, J.C.; Rich, M.M.; Maniatis, T. Distinct Roles for Motor Neuron Autophagy Early and Late in the SOD1 G93A Mouse Model of ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E8294–E8303.
- Hernandez, D.; Torres, C.A.; Setlik, W.; Cebrián, C.; Mosharov, E.V.; Tang, G.; Cheng, H.-C.; Kholodilov, N.; Yarygina, O.; Burke, R.E.; et al. Regulation of Presynaptic Neurotransmission by Macroautophagy. Neuron 2012, 74, 277–284.
- Binotti, B.; Pavlos, N.J.; Riedel, D.; Wenzel, D.; Vorbrüggen, G.; Schalk, A.M.; Kühnel, K.; Boyken, J.; Erck, C.; Martens, H.; et al. The GTPase Rab26 Links Synaptic Vesicles to the Autophagy Pathway. eLife 2015, 4, e05597.
- Nikoletopoulou, V.; Sidiropoulou, K.; Kallergi, E.; Dalezios, Y.; Tavernarakis, N. Modulation of Autophagy by BDNF Underlies Synaptic Plasticity. Cell Metab. 2017, 26, 230–242.e5.
- Yan, J.; Porch, M.W.; Court-Vazquez, B.; Bennett, M.V.L.; Zukin, R.S. Activation of Autophagy Rescues Synaptic and Cognitive Deficits in Fragile X Mice. Proc. Natl. Acad. Sci. USA 2018, 115, E9707–E9716.
- Tang, G.; Gudsnuk, K.; Kuo, S.-H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-Dependent Macroautophagy Causes Autistic-like Synaptic Pruning Deficits. Neuron 2014, 83, 1131–1143.
- Bateup, H.S.; Takasaki, K.T.; Saulnier, J.L.; Denefrio, C.L.; Sabatini, B.L. Loss of Tsc1 in Vivo Impairs Hippocampal mGluR-LTD and Increases Excitatory Synaptic Function. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 8862–8869.
- Amegandjin, C.A.; Choudhury, M.; Jadhav, V.; Carriço, J.N.; Quintal, A.; Berryer, M.; Snapyan, M.; Chattopadhyaya, B.; Saghatelyan, A.; Di Cristo, G. Sensitive Period for Rescuing Parvalbumin Interneurons Connectivity and Social Behavior Deficits Caused by TSC1 Loss. Nat. Commun. 2021, 12, 3653.
More