3. Drug-Related Factors
3.1. Pharmacokinetic Mechanisms
The metabolism of each generation of EGFR-TKIs is similar, but distinct. Based on its metabolism by CYP3A enzymes, most EGFR-TKIs, other than afatinib (second-generation), may be affected by CYP enzyme inducers, which might result in false resistance. For instance, dexamethasone, a CYP inducer, led to a 0.6-fold reduction in erlotinib (first-generation)
[17][24]. Other potent CYP inducers like phenytoin and rifampin can lower the level of gefitinib plasma concentrations
[18][19][25,26]. On the other hand, hepatic enzymes barely metabolize afatinib; hence, CYP-interacting substances are unlikely to have an effect on afatinib PK
[20][21][27,28].
3.2. Pharmacodynamic Mechanisms
This mechanism is mainly associated with intrinsic primary resistance. For instance, first- and second-generation EGFR-TKIs are ineffective against the
EGFR T790M mutation due to the consequent altered conformational structure of
EGFR. Thus, patients who had such a mutation originally are non-responders, known as those with intrinsic primary resistance.
Moreover, numerous studies indicate that responses to all-generation EGFR-TKIs vary among EGFR exon 20 mutations due to their heterogeneity
[22][23][31,32]. A recent study revealed that
EGFR mutations, encompassing atypical variations, can be organized into four unique subgroups based on their structures and functions to improve drug sensitivity prediction to targeted therapies: (1) classical-like mutations located away from the ATP-binding pocket; (2) T790M-like mutations within the hydrophobic core; (3) insertions in the loop at the C-terminal end of the αC-helix in exon 20 (Ex20ins-L); and (4) mutations on the inner surface of the ATP-binding pocket or at the C-terminal end of the αC-helix, anticipated to cause compression of P-loop and αC-helix (PACC)
[24][33]. Based on this structural classification, exon 20 mutations are sorted into one of three structural classes ((2), (3), and (4)). Especially among the (3) Ex20ins-L group, patients in the exon 20 far-loop insertions (Ex20ins-FL) subgroup are not sensitive to any targeted drugs, leading to intrinsic primary resistance.
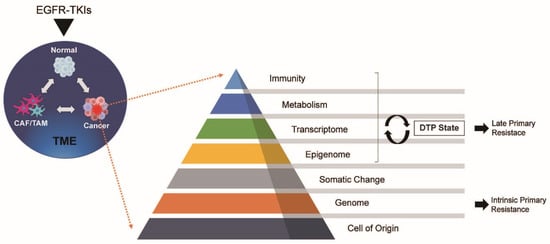
4. Tumor-Related Factors
4.1. Metabolism
4.1.1. DTP Cells and Late Primary Resistance
Cancer cells modify their metabolic processes to adjust to the changes brought about by drug exposure. For example, the Warburg Effect, proposed by Otto Warburg more than 50 years ago, is a phenomenon by which cancer cells harness anaerobic glycolysis to fuel their proliferation rather than mitochondrial oxidative phosphorylation
[25][34]. In contrast, recent research indicates that cancer cells use oxygen via mitochondrial oxidative phosphorylation as much as or more than normal cells
[26][35]. DTP cells, which are exposed by drugs, perform a pivotal role in this situation, leading to late primary resistance for EGFR-TKIs.
Since DTP cells demonstrate a heightened reliance on mitochondrial oxidative phosphorylation, they must regulate the overproduction of oxygen-rich molecules, such as superoxide and lipid peroxide, which arise from the mitochondrial oxidative phosphorylation process. Therefore, DTP cells use glutathione peroxidase 4 (GPX4) to neutralize lipid peroxides. It is confirmed in NSCLC cells that inhibiting GPX4 can lead to an accumulation of lipid peroxides, resulting in ferroptosis, a type of oxidative cell death
[27][28][36,37].
4.1.2. Lipid Metabolism and Autophagy
Lipid metabolism might also play a role in late primary resistance. Fatty acid β-oxidation is essential for energy generation in mitochondrial oxidative phosphorylation. CD36 acts as the primary transporter for fatty acid absorption. The high methylation of CD36 in lung cancer fills a vital role in cancer progression through fatty acid β-oxidation. As a matter of fact, de-methylation of CD36 can lead to removal of the resistance. Combined treatment with the DNA methylation inhibitor (decitabine) and the histone deacetylase (HDAC) inhibitor (chidamide) effectively curbs lung cancer growth in vivo
[29][39].
Autophagy serves as a cellular survival strategy by breaking down and recycling proteins, macromolecules, and organelles, in contrast to apoptosis, which leads to cell death. It plays a crucial role in clearing out malfunctioning mitochondria generated by reactive oxygen species during mitochondrial respiration. Drug-induced accumulation of these impaired mitochondria can enhance malignancy by amplifying oxidative stress. In a lung cancer mouse model, for example, removing the vital autophagy gene Atg7 has been shown to boost oxidative stress and speed up tumor cell growth
[30][31][40,41].
4.2. Transcriptome and Epigenome
Epigenetics encompasses gene regulatory processes that do not involve DNA sequences, including DNA methylation, hydroxymethylation, and modifications to histone proteins. The transcriptome is intricately linked to epigenetics. For instance, changes in chromatin structure facilitate the epigenetic control of apoptosis and gene expression. Some long noncoding RNAs are involved in oncogenesis through the regulation of transcription, translation, and epigenetics
[32][42].
A characteristic of DTP cells is their reversible biological ability, which is not determined by the genome, but rather by adaptable epigenomic regulation
[13][19].
Exposure to EGFR-TKIs can boost the expression of the epigenetic modulator KDM5A, which is an H3K4 demethylase. Given that KDM5A inhibits the transcriptional activity of H3K4, its increased expression can contribute to late primary resistance.
4.3. Somatic and Genomic Changes
4.3.1. TP53
p53 can induce cell cycle halting, senescence, and programmed cell death
[33][48]. Alterations in the TP53 gene, which codes for p53, are detected in 35–55% of NSCLC patients and are strongly linked to smoking behaviors
[34][49]. TP53 is the primary concurrent alteration in EGFR-mutant NSCLC patients, occurring in 55–65% of cases
[35][36][37][45,50,51].
The TP53 status correlates with reduced effectiveness to EGFR-TKIs. In NSCLC cell lines with wild-type p53, gefitinib can induce apoptosis by upregulating Fas at the plasma membrane and reviving caspase activation, thus increasing TKI responsiveness.
4.3.2. PIK3CA Mutation
PIK3CA is responsible for encoding the active component of PI3K, and its mutations can stimulate the PI3K/AKT pathway
[38][54]. Mutations in PIK3CA are uncommon, occurring in roughly 2–5% of NSCLC cases. The most frequent mutations are E545K in exon 9 and H1047R in exon 20
[39][40][41][55,56,57].
PIK3CA mutations frequently appear alongside other driver mutations, notably EGFR and KRAS
[39][42][43][55,58,59]. PI3KCA mutations are present in roughly 3.5% of patients with EGFR mutations and are associated with intrinsic primary resistance to EGFR-TKIs
[44][45][60,61].
4.3.3. PTEN Alterations
Phosphatase and tensin homologs deleted on chromosome 10 (PTEN) act as tumor suppressors and play a role in various cellular activities, including cell proliferation, survival, growth, metabolism, migration, and apoptosis
[46][47][48][64,65,66]. PTEN serves as a primary inhibitor of the PI3K/AKT pathway, and its inactivation is crucial for the onset and progression of lung cancer
[49][50][67,68]. Loss of PTEN function, found in more than 40% of cases, can activate the PI3K/AKT pathway, accelerating tumor progression and reducing sensitivity to EGFR-TKIs in NSCLC patients
[50][51][52][68,69,70]. PTEN mutations occur rarely, in 2–5% of NSCLC cases
[53][54][71,72].
This is due to PTEN’s role in regulating the endocytic trafficking of EGFR, a crucial mechanism for controlling EGFR signaling
[55][73]. Following the binding of EGF (ligand) to its receptor, EGFR, the receptor-ligand combination is taken up by clathrin-coated vesicles. These vesicles then transport the complex to early endosomes for categorization
[56][57][74,75]. The EGF/EGFR combination progresses to late endosomes via vesicle maturation and subsequently undergoes lysosomal fusion, leading to receptor degradation
[58][59][76,77]. PTEN plays a pivotal role in facilitating the transition of ligand-bound EGFR from early endosomes to late endosomes for receptor degradation by regulating the phosphorylation of Rab7 and maturing late endosomes
[60][78].
4.4. Cell of Origin
4.4.1. Lineage Plasticity
Histologic transformation is dominated by small-cell and squamous-cell lung cancer transformation as late primary resistance.
As for the mechanisms of SCLC transformation, there are two hypotheses. One theory is that NSCLC can be histologically differentiated into SCLC by EGFR-TKIs exposure through the inactivation of p53 and RB1
[61][79]. The other theory is that both SCLC and NSCLC components exist simultaneously in the same tumor. Since EGFR-TKIs can reduce the NSCLC elements of tumors, SCLC elements can remain and become dominant
[62][80].
4.4.2. BIM Deletion Polymorphism
BIM, also referred to as BCL2L11 or B-cell chronic lymphocytic leukemia/lymphoma-like 11, is a proapoptotic member of the Bcl-2 family that solely possesses BH3 domains. These domains of the BIM gene are essential for apoptosis triggered by EGFR-TKIs
[63][64][65][66][67][81,82,83,84,85]. Since BIM is degraded by ERK signaling, EGFR-TKIs can inhibit the ERK signaling and increase the level of BIM protein, leading to cell apoptosis.
The BIM deletion polymorphism involves a 2903 bp fragment removal in the BIM gene’s intron 2, producing an inactive BIM protein variant. This variant is missing the essential BH3 domain, compromising apoptosis related to EGFR-TKIs and leading to inherent primary resistance to these inhibitors
[64][82]. This BIM deletion polymorphism is found in 12–16% of EGFR-mutant lung cancer patients
[68][69][86,87], especially in East Asian individuals
[64][82]
5. Tumour Microenvironment (TME)
Exposure to drugs impacts the TME, which is linked to primary resistance against EGFR-TKIs. CAFs, a significant component of the TME, are known to contribute to drug resistance by releasing growth factors and chemokines
[70][91].
Additionally, TAMs and MDSCs play a crucial role in contributing to primary resistance to EGFR-TKIs. Specifically, MDSCs that are positive for S100A9, capable of differentiating into TAMs, have been linked to a diminished response to EGFR-TKIs in NSCLC patients
[71][94]. From a mechanistic perspective, S100A9 enhances ALDH1A1 expression and activates the retinoic acid (RA) signaling pathway, thereby promoting cancer proliferation. Using a pan-RAR antagonist can significantly reduce cancer growth
[72][95].
6. Summary
Removing the primary resistance for EGFR-TKIs has become a topic of increasing interest. Previous studies have focused on individual cancer cells, elucidating the acquired resistance mechanisms and investigating the treatment strategies. However, cancer cells interact with the surrounding environment and find another signal pathway for proliferation as a loophole. Tumors can progress on their own and be assisted by the surrounding environment.