Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Caroline Desmetz.
Fibrosis is a common feature of cardiovascular diseases and targets multiple organs, such as the heart and vessels. Endothelial to mesenchymal transition is a complex, vital process that occurs during embryonic formation and plays a crucial role in cardiac development. It is also a fundamental process implicated in cardiac fibrosis and repair, but also in other organs. Indeed, in numerous cardiovascular diseases, the endothelial-to-mesenchymal transition has been shown to be involved in the generation of fibroblasts that are able to produce extracellular matrix proteins such as type I collagen. This massive deposition results in tissue stiffening and organ dysfunction.
- fibrosis
- cardiovascular disease
- endothelial to mesenchymal transition
1. Introduction
Cardiovascular diseases (CVD) refer to a range of disorders that specifically impact the circulating system (heart and vessel network). According to the World Health Organization (WHO), CVD is a major cause of death, accounting for an estimated 17.9 million people every year and representing approximately 30% of deaths worldwide (WHO 2023). Myocardial fibrosis represents a dynamic restructuring of the extracellular matrix (ECM) in response to cardiac stress. Two types of mechanisms have been involved in cardiac fibrosis: “replacement” or “reparative” fibrosis is implicated in the reparative mechanisms that unfold during the acute phase of injuries, such as myocardial infarctions, pulmonary arterial hypertension or congenital heart disease, in order to locally replace dying cardiomyocytes [1]. In stark contrast, “reactive” or “diffuse” myocardial fibrosis corresponds to ECM remodeling developing gradually over time in response to chronic pressure overload and results in the progressive deposition of collagen in the interstitial and perivascular regions [1,2,3][1][2][3]. This persistent collagen deposition disrupts the normal function of either the left or the right ventricle of the heart, leading to ventricular stiffness and poor electrical conductance of the myocardium. Left ventricle reactive fibrosis occurs more specifically in chronic pathological conditions, such as systemic hypertension, aortic stenosis, diabetes mellitus [2], obesity [4], and cardiorenal syndrome [5] and is a key contributing factor leading to arrhythmias and progression to heart failure [1]. Therefore, investigation of the cellular and molecular mechanisms involved in the initiation and progression of myocardial fibrosis is of major importance to prevent its onset in high-risk but not yet diseased patients.
Endothelial to mesenchymal transition (EndMT) is a complex, vital process that occurs during embryonic formation and plays a crucial role in cardiogenesis, specifically in cardiac valve formation. This transdifferentiation process is characterized by the loss of endothelial fate and the acquisition of mesenchymal features, such as fibroblasts secreting extracellular matrix proteins. EndMT has emerged as a fundamental process implicated in cardiac fibrosis [6,7,8,9,10][6][7][8][9][10]. EndMT has also been shown to be involved in many cardiovascular diseases, including pulmonary arterial hypertension [7,8,11[7][8][11][12],12], atherosclerosis [13], diabetes mellitus [14,15[14][15][16][17],16,17], and chronic kidney disease [18]. However, the precise contribution of EndMT to cardiac fibrosis and CVD remains unclear, and efforts must be made to clarify those mechanisms. It is, therefore, essential to establish relevant cellular and animal models to elucidate mechanisms involved in this phenomenon.
2. Endothelial to Mesenchymal Transition in Cardiovascular Diseases
Endothelial-mesenchymal transition is a complex process that involves a transition from endothelial cells (ECs) towards a mesenchymal-like phenotype in response to a range of specific stimulators. Cells undergoing EndMT adopt a spindle-shaped morphology that facilitates their unrestricted movement within the ECM because of the loss of their cellular polarity and downregulation of adherent and tight junction proteins. This mobility enables them to assemble the connective tissue crucial for organ function [8]. EndMT mainly occurs during embryonic development, particularly in the early stages of cardiac septum and valve formation [8,9][8][9]. It is generally accepted that this transition is characterized by the loss of endothelial markers, such as platelet endothelial cell adhesion molecule (Pecam-1/CD31), vascular endothelial cadherin (VE-cadherin), von Willebrand factor (vWF), and the acquirement of a mesenchymal (fibroblastic-like) phenotype, characterized by the expression of mesenchymal markers including SM22-α (Smooth muscle protein 22-alpha), vimentin, α-SMA (Actin alpha 1 skeletal muscle), type I collagen, FSP-1 (Fibroblast specific protein-1, i.e., S100A4), fibronectin [6,8][6][8] (Figure 1A). Nevertheless, when examining EndMT at the molecular level, there is currently no consensus on criteria allowing the precise definition of this process. This lack of standardization poses a growing challenge, as it leads to a lack of uniformity and limited comparability of data issued from various model systems and research publications [9].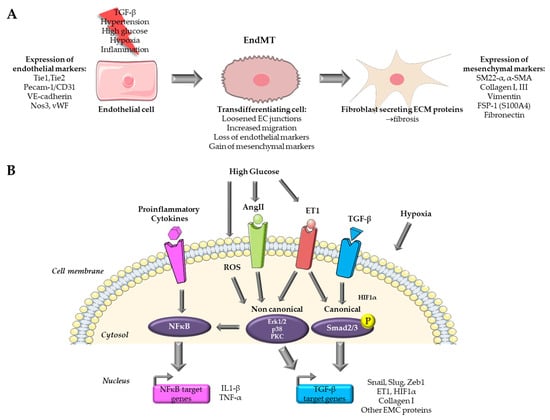
Figure 1. Overview of pathways targeted for the study of EndMT in cardiovascular diseases. (A) Endothelial to mesenchymal transition process. (B) Pathways targeted in in vitro models for the study of EndMT. Parts of the figure were drawn by using pictures from Servier Medical Art.
3. Existing In Vitro Models of Endothelial to Mesenchymal Transition in Cardiovascular Diseases
The majority of in vitro models for EndMT utilize primary ECs. In vitro models offer several advantages over their in vivo counterparts, including ease of accessibility, cost-effectiveness, and the potential for achieving highly reproducible outcomes. Nevertheless, it is important to acknowledge that in vitro models do have limitations when compared to in vivo models, and these limitations may impact the extrapolation of findings derived from such models. An overview of the signaling pathways targeted by in vitro models for the study of EndMT in cardiovascular diseases is presented in Figure 1B.3.1. Cytokine Based In Vitro Models
3.1.1. TGF-β Signaling
Among the growth factor families, TGF-β emerges as the most significant cytokine-inducing transdifferentiation of EC (recent review [19]). TGF-β plays a pivotal role at the early embryonic stages in guiding the atrioventricular canal formation [20]. TGF-β is secreted by both fibroblasts and macrophages and stimulates collagen and ECM component production by enhancing EndMT, which in turn leads to fibrotic lesions [21]. The initiation of TGF-β-induced EndMT signalization following the canonical pathway is achieved through the binding of TGF-β isoforms to its receptor TGFBR1/2, activating its intrinsic kinase activity. This cascade of kinase/phosphorylation activity continues within the cell to the nucleus, across phosphorylation of downstream effectors, including receptor-activated Smad (R-Smads) and particularly Smad2/3, leading to the formation of a hetero-oligomeric complex with Smad4. This promotes their translocation in the nucleus where the complex binds to target gene promoters, inducing the expression of transcription factors like Snail, Slug, and Twist, which leads to the up-regulation of numerous EndMT-involved genes [8,19][8][19]. As for the canonical pathway, emerging evidence highlights the involvement of non-canonical pathways in the signaling of TGF-β-induced EndMT through the activation of a Smad-independent kinase complex, which requires several other downstream effectors, such as the mitogen-activated protein kinase (MAPK), PI3K, and PKC-δ. Indeed, TGF-β has been shown to induce the activation of MAPK cascades in a fibrosis context, triggering extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 MAPK. These activated downstream effectors can transduce signals to the nucleus, leading to an increased expression of transcription factors, thereby stimulating the up-regulation of EndMT-associated gene expression [19]. While the precise mechanisms governing these pathways exhibit diversity and remain partially understood, the majority of these pathways ultimately intersect by modulating transcription factors, such as Snail and Twist, to inhibit the expression of EC markers and to induce the expression of mesenchymal proteins.3.1.2. TGF-β Isoform Specific Effects to Induce EndMT
Varying degrees of potency in promoting EndMT has been attributed to the three TGF-β isoforms, namely TGF-β1, TGF-β2, and TGF-β3. Among them, TGF-β2 appears to be the isoform with the greatest potential for inducing EndMT, as highlighted during embryonic heart development [22] and on immortalized human dermal microvascular endothelial cells (HMVEC) [23,24][23][24]. Exposure for 72 h to 1 ng/mL TGF-β1 and TGF-β3 led to an increase in the expression of TGF-β2, while TGF-β2 treatment did not. This suggests that EndMT induced by TGF-β1 and TGF-β3 is the result of secondary effects mediated through the secretion of TGF-β2. Moreover, TGF-β2 increases phosphorylation (activation) of Smad2/3 (canonical) and p38 MAPK (non-canonical) at a higher level than TGF-β1 and TGF-β3. Finally, silencing TGF-β2 attenuated the expression of EndMT markers in cells treated with TGF-β1 and TGF-β3. This, therefore, suggests that TGF-β1 and TGF-β3-induced EndMT involves a paracrine loop mediated by TGF-β2 [23]. Interestingly, studies carried out over the last 10 years have used either TGF-β2 [23,24,25,26][23][24][25][26] or TGF-β1 on endothelial cells from different origins [27,28,29,30,31,32,33,34][27][28][29][30][31][32][33][34] and, more rarely, TGF-β3 [23,35][23][35].3.1.3. EndMT Induction Level Depend on Endothelial Cell Origin
Most in vitro models use endothelial cells of different anatomical origins treated with a member of the TGF-β family. In these models, EndMT is characterized by four characteristics: (1) simultaneous expression of endothelial and mesenchymal markers, (2) heightened cellular migratory capabilities, (3) diminished expression of endothelial traits, including reduced leukocyte adhesion and impaired tubule formation, and (4) increased manifestation of mesenchymal/myofibroblastic traits, such as augmented collagen production and enhanced contractility [36]. Several EC models are widely used, such as human umbilical vein ECs (HUVEC), aortic EC (HAEC), coronary EC (coronary artery CAEC, or microvascular CMVEC), cardiac EC (microvascular CMEC, atrial endocardial AEEC), pulmonary artery EC (PAEC), and dermal EC (HMEC). Two studies provide interesting evidence that cells from different anatomical origins do not have the same potential to enter transdifferentiation. Among the origins tested (coronary artery, aortic, umbilical vein, pulmonary artery), cells of aortic and coronary origin showed the best responses to induction by TGF-β2 treatment. This results in an increase in several mesenchymal markers, such as type I collagen, actin alpha cardiac muscle (ACTC), SM22-α, Calponin 1 (CNN1), Snail, and maintenance of endothelial markers such as Pecam-1 and VE-cadherin. This is also accompanied by a morphological and functional change, with a diminished ability to form vessel-like structures [25]. In cells of aortic origin, the induction of EndMT is accompanied by significant activation of the non-canonical pathway via ERK1/2. In aortic and coronary cells, induction is potentiated by Snail overexpression in combination with TGF-β2 treatment, which is less obvious for HUVEC [37] or PAEC [25,37,38][25][37][38]. Moreover, cells of umbilical and pulmonary origin showed a delay or resistance to enter transdifferentiation and do not produce type I collagen (something also observed in the laboratory, unpublished data). It is, therefore, clear that the origin of EC plays a role in their ability to enter EndMT and that more information is required about these origins, as well as the conditions under which EndMT is induced.3.1.4. Induction of EndMT by Co-Treatment with Proinflammatory Signals
The second most widely used model is that of EC treated with a combination of TGF-β and a proinflammatory cytokine, like IL1-β, TNF-α, or both. Indeed, the inflammatory context found in many cardiovascular diseases has been shown to promote cardiac fibrosis [3]. Most studies demonstrate that induction of EndMT is possible using TGF-β isoforms in combination with a proinflammatory cytokine [35,38,39][35][38][39]. This is the case for HUVEC, where costimulation with IL-1β and TGF-β1 or 2 caused synergistic induction of EndMT accompanied by overexpression of the transcription factor NF-κB, an increase in its nuclear translocation [40,41][40][41], and type I collagen expression, which was also the case in PAEC [32,38][32][38]. Cotreatment with TNF-α also induced EndMT [42]. Treatment with IL-1β and TNF-α without TGF-β was also able to induce expression changes related to EndMT in HUVEC [41,43][41][43], suggesting that these cells may be more susceptible to inflammatory stimuli. This was also demonstrated in other organ fibrosis disorders, such as intestinal fibrosis [44]. However, in that particular case, no significant change in their migratory capacity was observed, confirming a resistant or delayed capacity of entering EndMT in HUVEC and PAEC [38], showing once again that cell origin plays a role in the ability to enter EndMT. Finally, angiotensin II (Ang II), a major fibrosis inducer [3], is also able to induce EndMT by activating NFκB signaling and pro-inflammatory cytokine production in HUVEC [45,46,47,48][45][46][47][48] in HMVEC [49] and in HCAEC [50]. AngII also induces and activates the TGF-β axis [3]. In short, proinflammatory signals, whether or not associated with TGF-β, are capable of inducing EndMT in ECs and represent widely used in vitro models.3.2. Hypoxia-Based In Vitro Models
Chronic hypoxia plays a pivotal role in the development of cardiac fibrosis and is induced by a reduced density of microvessels, which subsequently impacts oxygen delivery and increased oxygen consumption due to the activation of inflammatory cells and fibroblasts. Chronic hypoxia independently contributes to abnormal ventricular remodeling and the onset of cardiac fibrosis [51]. Hypoxia has the capability to trigger EndMT via HIF1 signaling, even in the absence of TGF-β, as it directly regulates Snail expression through HIF1α in HCAEC [52]. Several publications, such as MVEC [53], HCAEC [52], and HUVEC [54[54][55][56],55,56], use cells under hypoxia as an EndMT model. Interestingly, hypoxia is widely used on PAEC [57,58,59][57][58][59] and PMVEC of pulmonary origin [60]. Indeed, extensive vascular remodeling occurs in pulmonary arterial hypertension (PAH), where neointimal and medial thickening results from fibrosis in the pulmonary arteries [12]. In vitro models of hypoxia-induced EndMT were developed in the last 10 years and allowed elucidation of signaling cascades activated in response to hypoxia in PAH (reviewed in [12]). Hypoxia conditioning of EC, therefore, appears to be a relevant in vitro model for studying EndMT, especially in PAH.3.3. High Glucose Based In Vitro Models of Organ Fibrosis in Diabetes Mellitus
One of the consequences of hyperglycemia is vascular dysfunction, which is a main component of diabetes mellitus, leading to both micro- and macro-vascular complications, and which affects several organs such as the kidney (diabetic nephropathy), heart (diabetic cardiomyopathy), retina (diabetic retinopathy), resulting in organ fibrosis. EndMT has been identified as an early important potential trigger in diabetic complications, as ECs are among the first to be damaged by hyperglycemia (recent review in [17]). High glucose activates a variety of EndMT-inducing pathways, such as TGF-β (through the PKC pathway), ET-1, and inflammatory signals. EndMT models were therefore designed with EC from these different origins and treated with high glucose concentrations in order to mimic hyperglycemia. HUVEC is widely used as a model for diabetes-associated cardiac fibrosis [14,61,62,63,64,65,66,67,68,69,70][14][61][62][63][64][65][66][67][68][69][70]. In particular, Yu et al. showed that 24 h treatment with 30 mM glucose resulted in the expression of the mesenchymal markers α-SMA and type I collagen in HUVEC. Endothelial markers (Pecam-1, VE-cadherin) were under-expressed more slowly or at higher glucose concentrations, reflecting a delay in the regulation of these markers during glucose-mediated EndMT. Finally, mesenchymal cells were obtained at 60 mM glucose and after 48 h of treatment. High glucose-induced TGF-β1 production from 48 h of treatment [63] shows that high glucose-mediated EndMT involves the TGF-β pathway. In vitro studies using HUVEC are usually combined with in vivo studies on animal models of diabetic disease. Aortic EC and CAEC are also used to study diabetic cardiomyopathy [71,72,73,74,75][71][72][73][74][75]. It is worth noting that to study EndMT in diabetic nephropathy and kidney fibrosis, glomerular endothelial cells (GEnC) are specifically used [76,77,78,79,80,81,82,83][76][77][78][79][80][81][82][83]. Finally, human retinal EC is used to study diabetic retinopathy [84,85,86,87,88][84][85][86][87][88]. Recently, a new 3D model of the aortic valve was developed in order to enhance the comprehension of the dynamic interplay between ECs and their microenvironmental matrix in the context of hyperglycemia [89]. The authors used hydrogel containing extracellular matrix from porcine aortic root and human valve cells. They cultivated valve EC on the hydrogel’s surface and valve interstitial cells within the hydrogel, then exposed this 3D structure to high glucose conditions. The authors observed a reduced expression of endothelial markers Pecam-1 and VE-cadherin and increased expression of mesenchymal markers α-SMA and Vimentin. There was also a loss of intercellular junctions and an enhanced expression of inflammatory molecules. Finally, valvular ECs showed enhanced monocyte adhesion via a mechanism involving adhesion molecules such as ICAM-1 and VCAM-1, showing dysfunctionality. The 3D model contains an ECM mainly composed of collagen I and III, similar to the aortic valve structure [89]. This model could also be interesting for cardiac myocardial fibrosis because ECM has a similar composition.References
- Schimmel, K.; Ichimura, K.; Reddy, S.; Haddad, F.; Spiekerkoetter, E. Cardiac Fibrosis in the Pressure Overloaded Left and Right Ventricle as a Therapeutic Target. Front. Cardiovasc. Med. 2022, 9, 886553.
- Maruyama, K.; Imanaka-Yoshida, K. The Pathogenesis of Cardiac Fibrosis: A Review of Recent Progress. Int. J. Mol. Sci. 2022, 23, 2617.
- Frangogiannis, N.G. Cardiac Fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488.
- Kruszewska, J.; Cudnoch-Jedrzejewska, A.; Czarzasta, K. Remodeling and Fibrosis of the Cardiac Muscle in the Course of Obesity-Pathogenesis and Involvement of the Extracellular Matrix. Int. J. Mol. Sci. 2022, 23, 4195.
- Valero-Muñoz, M.; Oh, A.; Faudoa, E.; Bretón-Romero, R.; El Adili, F.; Bujor, A.; Sam, F. Endothelial-Mesenchymal Transition in Heart Failure with a Preserved Ejection Fraction: Insights into the Cardiorenal Syndrome. Circ. Heart Fail. 2021, 14, e008372.
- Sun, X.; Nkennor, B.; Mastikhina, O.; Soon, K.; Nunes, S.S. Endothelium-Mediated Contributions to Fibrosis. Semin. Cell Dev. Biol. 2020, 101, 78–86.
- Anbara, T.; Sharifi, M.; Aboutaleb, N. Endothelial to Mesenchymal Transition in the Cardiogenesis and Cardiovascular Diseases. Curr. Cardiol. Rev. 2020, 16, 306–314.
- Peng, Q.; Shan, D.; Cui, K.; Li, K.; Zhu, B.; Wu, H.; Wang, B.; Wong, S.; Norton, V.; Dong, Y.; et al. The Role of Endothelial-to-Mesenchymal Transition in Cardiovascular Disease. Cells 2022, 11, 1834.
- Kovacic, J.C.; Dimmeler, S.; Harvey, R.P.; Finkel, T.; Aikawa, E.; Krenning, G.; Baker, A.H. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 190–209.
- Hong, L.; Du, X.; Li, W.; Mao, Y.; Sun, L.; Li, X. EndMT: A Promising and Controversial Field. Eur. J. Cell Biol. 2018, 97, 493–500.
- Gaikwad, A.V.; Eapen, M.S.; McAlinden, K.D.; Chia, C.; Larby, J.; Myers, S.; Dey, S.; Haug, G.; Markos, J.; Glanville, A.R.; et al. Endothelial to Mesenchymal Transition (EndMT) and Vascular Remodeling in Pulmonary Hypertension and Idiopathic Pulmonary Fibrosis. Expert. Rev. Respir. Med. 2020, 14, 1027–1043.
- Gorelova, A.; Berman, M.; Al Ghouleh, I. Endothelial-to-Mesenchymal Transition in Pulmonary Arterial Hypertension. Antioxid. Redox Signal. 2021, 34, 891–914.
- Souilhol, C.; Harmsen, M.C.; Evans, P.C.; Krenning, G. Endothelial-Mesenchymal Transition in Atherosclerosis. Cardiovasc. Res. 2018, 114, 565–577.
- Widyantoro, B.; Emoto, N.; Nakayama, K.; Anggrahini, D.W.; Adiarto, S.; Iwasa, N.; Yagi, K.; Miyagawa, K.; Rikitake, Y.; Suzuki, T.; et al. Endothelial Cell-Derived Endothelin-1 Promotes Cardiac Fibrosis in Diabetic Hearts through Stimulation of Endothelial-to-Mesenchymal Transition. Circulation 2010, 121, 2407–2418.
- Tang, R.-N.; Lv, L.-L.; Zhang, J.-D.; Dai, H.-Y.; Li, Q.; Zheng, M.; Ni, J.; Ma, K.-L.; Liu, B.-C. Effects of Angiotensin II Receptor Blocker on Myocardial Endothelial-to-Mesenchymal Transition in Diabetic Rats. Int. J. Cardiol. 2013, 162, 92–99.
- Murdoch, C.E.; Chaubey, S.; Zeng, L.; Yu, B.; Ivetic, A.; Walker, S.J.; Vanhoutte, D.; Heymans, S.; Grieve, D.J.; Cave, A.C.; et al. Endothelial NADPH Oxidase-2 Promotes Interstitial Cardiac Fibrosis and Diastolic Dysfunction through Proinflammatory Effects and Endothelial-Mesenchymal Transition. J. Am. Coll. Cardiol. 2014, 63, 2734–2741.
- Wang, E.; Wang, H.; Chakrabarti, S. Endothelial-to-Mesenchymal Transition: An Underappreciated Mediator of Diabetic Complications. Front. Endocrinol. 2023, 14, 1050540.
- Charytan, D.M.; Padera, R.; Helfand, A.M.; Zeisberg, M.; Xu, X.; Liu, X.; Himmelfarb, J.; Cinelli, A.; Kalluri, R.; Zeisberg, E.M. Increased Concentration of Circulating Angiogenesis and Nitric Oxide Inhibitors Induces Endothelial to Mesenchymal Transition and Myocardial Fibrosis in Patients with Chronic Kidney Disease. Int. J. Cardiol. 2014, 176, 99–109.
- Massagué, J.; Sheppard, D. TGF-β Signaling in Health and Disease. Cell 2023, 186, 4007–4037.
- Goumans, M.-J.; Ten Dijke, P. TGF-β Signaling in Control of Cardiovascular Function. Cold Spring Harb. Perspect. Biol. 2018, 10, a022210.
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β Signaling in Fibrosis. Growth Factors 2011, 29, 196–202.
- Azhar, M.; Runyan, R.B.; Gard, C.; Sanford, L.P.; Miller, M.L.; Andringa, A.; Pawlowski, S.; Rajan, S.; Doetschman, T. Ligand-Specific Function of Transforming Growth Factor Beta in Epithelial-Mesenchymal Transition in Heart Development. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2009, 238, 431–442.
- Sabbineni, H.; Verma, A.; Somanath, P.R. Isoform-Specific Effects of Transforming Growth Factor β on Endothelial-to-Mesenchymal Transition. J. Cell. Physiol. 2018, 233, 8418–8428.
- Medici, D.; Potenta, S.; Kalluri, R. Transforming Growth Factor-Β2 Promotes Snail-Mediated Endothelial-Mesenchymal Transition through Convergence of Smad-Dependent and Smad-Independent Signalling. Biochem. J. 2011, 437, 515–520.
- Ursoli Ferreira, F.; Eduardo Botelho Souza, L.; Hassibe Thomé, C.; Tomazini Pinto, M.; Origassa, C.; Salustiano, S.; Marcel Faça, V.; Olsen Câmara, N.; Kashima, S.; Tadeu Covas, D. Endothelial Cells Tissue-Specific Origins Affects Their Responsiveness to TGF-Β2 during Endothelial-to-Mesenchymal Transition. Int. J. Mol. Sci. 2019, 20, 458.
- Pinto, M.T.; Covas, D.T.; Kashima, S.; Rodrigues, C.O. Endothelial Mesenchymal Transition: Comparative Analysis of Different Induction Methods. Biol. Proced. Online 2016, 18, 10.
- Wang, H.; Zhang, X.; Liu, Y.; Zhang, Y.; Wang, Y.; Peng, Y.; Ding, Y. Diosmetin-7-O-β-D-Glucopyranoside Suppresses Endothelial-Mesenchymal Transformation through Endoplasmic Reticulum Stress in Cardiac Fibrosis. Clin. Exp. Pharmacol. Physiol. 2023, 50, 789–805.
- Wang, B.; Ge, Z.; Wu, Y.; Zha, Y.; Zhang, X.; Yan, Y.; Xie, Y. MFGE8 Is Down-Regulated in Cardiac Fibrosis and Attenuates Endothelial-Mesenchymal Transition through Smad2/3-Snail Signalling Pathway. J. Cell. Mol. Med. 2020, 24, 12799–12812.
- Lai, Y.-J.; Tsai, F.-C.; Chang, G.-J.; Chang, S.-H.; Huang, C.-C.; Chen, W.-J.; Yeh, Y.-H. miR-181b Targets Semaphorin 3A to Mediate TGF-β-Induced Endothelial-Mesenchymal Transition Related to Atrial Fibrillation. J. Clin. Investig. 2022, 132, e142548.
- Wilhelmi, T.; Xu, X.; Tan, X.; Hulshoff, M.S.; Maamari, S.; Sossalla, S.; Zeisberg, M.; Zeisberg, E.M. Serelaxin Alleviates Cardiac Fibrosis through Inhibiting Endothelial-to-Mesenchymal Transition via RXFP1. Theranostics 2020, 10, 3905–3924.
- Chen, H.; Liu, Y.; Gui, Q.; Zhu, X.; Zeng, L.; Meng, J.; Qing, J.; Gao, L.; Jackson, A.O.; Feng, J.; et al. Ghrelin Attenuates Myocardial Fibrosis after Acute Myocardial Infarction via Inhibiting Endothelial-to Mesenchymal Transition in Rat Model. Peptides 2019, 111, 118–126.
- Rinastiti, P.; Ikeda, K.; Rahardini, E.P.; Miyagawa, K.; Tamada, N.; Kuribayashi, Y.; Hirata, K.-I.; Emoto, N. Loss of Family with Sequence Similarity 13, Member A Exacerbates Pulmonary Hypertension through Accelerating Endothelial-to-Mesenchymal Transition. PLoS ONE 2020, 15, e0226049.
- Song, S.; Liu, L.; Yu, Y.; Zhang, R.; Li, Y.; Cao, W.; Xiao, Y.; Fang, G.; Li, Z.; Wang, X.; et al. Inhibition of BRD4 Attenuates Transverse Aortic Constriction- and TGF-β-Induced Endothelial-Mesenchymal Transition and Cardiac Fibrosis. J. Mol. Cell. Cardiol. 2019, 127, 83–96.
- Zhang, H.; Hu, J.; Liu, L. MiR-200a Modulates TGF-Β1-Induced Endothelial-to-Mesenchymal Shift via Suppression of GRB2 in HAECs. Biomed. Pharmacother. 2017, 95, 215–222.
- Sánchez-Duffhues, G.; García de Vinuesa, A.; van de Pol, V.; Geerts, M.E.; de Vries, M.R.; Janson, S.G.; van Dam, H.; Lindeman, J.H.; Goumans, M.-J.; Ten Dijke, P. Inflammation Induces Endothelial-to-Mesenchymal Transition and Promotes Vascular Calcification through Downregulation of BMPR2. J. Pathol. 2019, 247, 333–346.
- Alvandi, Z.; Bischoff, J. Endothelial-Mesenchymal Transition in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2357–2369.
- Pinto, M.T.; Ferreira Melo, F.U.; Malta, T.M.; Rodrigues, E.S.; Plaça, J.R.; Silva, W.A.; Panepucci, R.A.; Covas, D.T.; de Oliveira Rodrigues, C.; Kashima, S. Endothelial Cells from Different Anatomical Origin Have Distinct Responses during SNAIL/TGF-Β2-Mediated Endothelial-Mesenchymal Transition. Am. J. Transl. Res. 2018, 10, 4065–4081.
- Monteiro, J.P.; Rodor, J.; Caudrillier, A.; Scanlon, J.P.; Spiroski, A.-M.; Dudnakova, T.; Pflüger-Müller, B.; Shmakova, A.; von Kriegsheim, A.; Deng, L.; et al. MIR503HG Loss Promotes Endothelial-to-Mesenchymal Transition in Vascular Disease. Circ. Res. 2021, 128, 1173–1190.
- Yoshimatsu, Y.; Wakabayashi, I.; Kimuro, S.; Takahashi, N.; Takahashi, K.; Kobayashi, M.; Maishi, N.; Podyma-Inoue, K.A.; Hida, K.; Miyazono, K.; et al. TNF-α Enhances TGF-β-Induced Endothelial-to-Mesenchymal Transition via TGF-β Signal Augmentation. Cancer Sci. 2020, 111, 2385–2399.
- Maleszewska, M.; Moonen, J.-R.A.J.; Huijkman, N.; van de Sluis, B.; Krenning, G.; Harmsen, M.C. IL-1β and TGFβ2 Synergistically Induce Endothelial to Mesenchymal Transition in an NFκB-Dependent Manner. Immunobiology 2013, 218, 443–454.
- Kim, S.; Lee, H.; Moon, H.; Kim, R.; Kim, M.; Jeong, S.; Kim, H.; Kim, S.H.; Hwang, S.S.; Lee, M.Y.; et al. Epigallocatechin-3-Gallate Attenuates Myocardial Dysfunction via Inhibition of Endothelial-to-Mesenchymal Transition. Antioxidants 2023, 12, 1059.
- Chen, D.; Zhang, C.; Chen, J.; Yang, M.; Afzal, T.A.; An, W.; Maguire, E.M.; He, S.; Luo, J.; Wang, X.; et al. miRNA-200c-3p Promotes Endothelial to Mesenchymal Transition and Neointimal Hyperplasia in Artery Bypass Grafts. J. Pathol. 2021, 253, 209–224.
- Miscianinov, V.; Martello, A.; Rose, L.; Parish, E.; Cathcart, B.; Mitić, T.; Gray, G.A.; Meloni, M.; Al Haj Zen, A.; Caporali, A. MicroRNA-148b Targets the TGF-β Pathway to Regulate Angiogenesis and Endothelial-to-Mesenchymal Transition during Skin Wound Healing. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1996–2007.
- Rieder, F.; Kessler, S.P.; West, G.A.; Bhilocha, S.; de la Motte, C.; Sadler, T.M.; Gopalan, B.; Stylianou, E.; Fiocchi, C. Inflammation-Induced Endothelial-to-Mesenchymal Transition: A Novel Mechanism of Intestinal Fibrosis. Am. J. Pathol. 2011, 179, 2660–2673.
- Lin, K.; Luo, W.; Yan, J.; Shen, S.; Shen, Q.; Wang, J.; Guan, X.; Wu, G.; Huang, W.; Liang, G. TLR2 Regulates Angiotensin II-Induced Vascular Remodeling and EndMT through NF-κB Signaling. Aging 2020, 13, 2553–2574.
- Jordan, N.P.; Tingle, S.J.; Shuttleworth, V.G.; Cooke, K.; Redgrave, R.E.; Singh, E.; Glover, E.K.; Ahmad Tajuddin, H.B.; Kirby, J.A.; Arthur, H.M.; et al. MiR-126-3p Is Dynamically Regulated in Endothelial-to-Mesenchymal Transition during Fibrosis. Int. J. Mol. Sci. 2021, 22, 8629.
- Zhang, Z.-Y.; Zhai, C.; Yang, X.-Y.; Li, H.-B.; Wu, L.-L.; Li, L. Knockdown of CD146 Promotes Endothelial-to-Mesenchymal Transition via Wnt/β-Catenin Pathway. PLoS ONE 2022, 17, e0273542.
- Krishnamoorthi, M.K.; Thandavarayan, R.A.; Youker, K.A.; Bhimaraj, A. An In Vitro Platform to Study Reversible Endothelial-to-Mesenchymal Transition. Front. Pharmacol. 2022, 13, 912660.
- Li, Z.; Kong, X.; Zhang, Y.; Zhang, Y.; Yu, L.; Guo, J.; Xu, Y. Dual Roles of Chromatin Remodeling Protein BRG1 in Angiotensin II-Induced Endothelial-Mesenchymal Transition. Cell Death Dis. 2020, 11, 549.
- Wang, J.; Li, H.; Lv, Z.; Luo, X.; Deng, W.; Zou, T.; Zhang, Y.; Sang, W.; Wang, X. The miR-214-3p/c-Ski Axis Modulates Endothelial-Mesenchymal Transition in Human Coronary Artery Endothelial Cells in Vitro and in Mice Model in Vivo. Hum. Cell 2022, 35, 486–497.
- Watson, C.J.; Collier, P.; Tea, I.; Neary, R.; Watson, J.A.; Robinson, C.; Phelan, D.; Ledwidge, M.T.; McDonald, K.M.; McCann, A.; et al. Hypoxia-Induced Epigenetic Modifications Are Associated with Cardiac Tissue Fibrosis and the Development of a Myofibroblast-like Phenotype. Hum. Mol. Genet. 2014, 23, 2176–2188.
- Xu, X.; Tan, X.; Tampe, B.; Sanchez, E.; Zeisberg, M.; Zeisberg, E.M. Snail Is a Direct Target of Hypoxia-Inducible Factor 1α (HIF1α) in Hypoxia-Induced Endothelial to Mesenchymal Transition of Human Coronary Endothelial Cells. J. Biol. Chem. 2015, 290, 16653–16664.
- Sniegon, I.; Prieß, M.; Heger, J.; Schulz, R.; Euler, G. Endothelial Mesenchymal Transition in Hypoxic Microvascular Endothelial Cells and Paracrine Induction of Cardiomyocyte Apoptosis Are Mediated via TGFβ1/SMAD Signaling. Int. J. Mol. Sci. 2017, 18, 2290.
- Fan, M.; Yang, K.; Wang, X.; Chen, L.; Gill, P.S.; Ha, T.; Liu, L.; Lewis, N.H.; Williams, D.L.; Li, C. Lactate Promotes Endothelial-to-Mesenchymal Transition via Snail1 Lactylation after Myocardial Infarction. Sci. Adv. 2023, 9, eadc9465.
- Helmke, A.; Casper, J.; Nordlohne, J.; David, S.; Haller, H.; Zeisberg, E.M.; von Vietinghoff, S. Endothelial-to-Mesenchymal Transition Shapes the Atherosclerotic Plaque and Modulates Macrophage Function. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 2278–2289.
- Zhang, G.-H.; Yu, F.-C.; Li, Y.; Wei, Q.; Song, S.-S.; Zhou, F.-P.; Tong, J.-Y. Prolyl 4-Hydroxylase Domain Protein 3 Overexpression Improved Obstructive Sleep Apnea-Induced Cardiac Perivascular Fibrosis Partially by Suppressing Endothelial-to-Mesenchymal Transition. J. Am. Heart Assoc. 2017, 6, e006680.
- Zhang, H.; Liu, Y.; Yan, L.; Du, W.; Zhang, X.; Zhang, M.; Chen, H.; Zhang, Y.; Zhou, J.; Sun, H.; et al. Bone Morphogenetic Protein-7 Inhibits Endothelial-Mesenchymal Transition in Pulmonary Artery Endothelial Cell under Hypoxia. J. Cell. Physiol. 2018, 233, 4077–4090.
- Song, S.; Zhang, M.; Yi, Z.; Zhang, H.; Shen, T.; Yu, X.; Zhang, C.; Zheng, X.; Yu, L.; Ma, C.; et al. The Role of PDGF-B/TGF-Β1/Neprilysin Network in Regulating Endothelial-to-Mesenchymal Transition in Pulmonary Artery Remodeling. Cell. Signal. 2016, 28, 1489–1501.
- Woo, K.V.; Shen, I.Y.; Weinheimer, C.J.; Kovacs, A.; Nigro, J.; Lin, C.-Y.; Chakinala, M.; Byers, D.E.; Ornitz, D.M. Endothelial FGF Signaling Is Protective in Hypoxia-Induced Pulmonary Hypertension. J. Clin. Investig. 2021, 131, e141467.
- Zhang, B.; Niu, W.; Dong, H.-Y.; Liu, M.-L.; Luo, Y.; Li, Z.-C. Hypoxia Induces Endothelial-mesenchymal Transition in Pulmonary Vascular Remodeling. Int. J. Mol. Med. 2018, 42, 270–278.
- Chen, Y.; Yang, Q.; Zhan, Y.; Ke, J.; Lv, P.; Huang, J. The Role of miR-328 in High Glucose-Induced Endothelial-to-Mesenchymal Transition in Human Umbilical Vein Endothelial Cells. Life Sci. 2018, 207, 110–116.
- Chen, X.-Y.; Lv, R.-J.; Zhang, W.; Yan, Y.-G.; Li, P.; Dong, W.-Q.; Liu, X.; Liang, E.-S.; Tian, H.-L.; Lu, Q.-H.; et al. Inhibition of Myocyte-Specific Enhancer Factor 2A Improved Diabetic Cardiac Fibrosis Partially by Regulating Endothelial-to-Mesenchymal Transition. Oncotarget 2016, 7, 31053–31066.
- Yu, C.-H.; Suriguga; Gong, M.; Liu, W.-J.; Cui, N.-X.; Wang, Y.; Du, X.; Yi, Z.-C. High Glucose Induced Endothelial to Mesenchymal Transition in Human Umbilical Vein Endothelial Cell. Exp. Mol. Pathol. 2017, 102, 377–383.
- Tsai, T.-H.; Lee, C.-H.; Cheng, C.-I.; Fang, Y.-N.; Chung, S.-Y.; Chen, S.-M.; Lin, C.-J.; Wu, C.-J.; Hang, C.-L.; Chen, W.-Y. Liraglutide Inhibits Endothelial-to-Mesenchymal Transition and Attenuates Neointima Formation after Endovascular Injury in Streptozotocin-Induced Diabetic Mice. Cells 2019, 8, 589.
- Li, Q.; Yao, Y.; Shi, S.; Zhou, M.; Zhou, Y.; Wang, M.; Chiu, J.-J.; Huang, Z.; Zhang, W.; Liu, M.; et al. Inhibition of miR-21 Alleviated Cardiac Perivascular Fibrosis via Repressing EndMT in T1DM. J. Cell. Mol. Med. 2020, 24, 910–920.
- Tian, J.; Zhang, M.; Suo, M.; Liu, D.; Wang, X.; Liu, M.; Pan, J.; Jin, T.; An, F. Dapagliflozin Alleviates Cardiac Fibrosis through Suppressing EndMT and Fibroblast Activation via AMPKα/TGF-β/Smad Signalling in Type 2 Diabetic Rats. J. Cell. Mol. Med. 2021, 25, 7642–7659.
- Lu, L.; Zhong, Z.; Gu, J.; Nan, K.; Zhu, M.; Miao, C. Ets1 Associates with KMT5A to Participate in High Glucose-Mediated EndMT via Upregulation of PFN2 Expression in Diabetic Nephropathy. Mol. Med. 2021, 27, 74.
- Ding, H.; Yao, J.; Xie, H.; Wang, C.; Chen, J.; Wei, K.; Ji, Y.; Liu, L. MicroRNA-195-5p Downregulation Inhibits Endothelial Mesenchymal Transition and Myocardial Fibrosis in Diabetic Cardiomyopathy by Targeting Smad7 and Inhibiting Transforming Growth Factor Beta 1-Smads-Snail Pathway. Front. Physiol. 2021, 12, 709123.
- Meng, Z.; Shen, W.; Yu, L.; Tong, F.; He, H.; Hu, Y.; Wu, W.; Liu, J. Bach1 Modulates AKT3 Transcription to Participate in Hyperglycaemia-Mediated EndMT in Vascular Endothelial Cells. Clin. Exp. Pharmacol. Physiol. 2023, 50, 443–452.
- Hulshoff, M.S.; Schellinger, I.N.; Xu, X.; Fledderus, J.; Rath, S.K.; Wong, F.C.; Maamari, S.; Haunschild, J.; Krenning, G.; Raaz, U.; et al. miR-132-3p and KLF7 as Novel Regulators of Aortic Stiffening-Associated EndMT in Type 2 Diabetes Mellitus. Diabetol. Metab. Syndr. 2023, 15, 11.
- Zhu, G.-H.; Li, R.; Zeng, Y.; Zhou, T.; Xiong, F.; Zhu, M. MicroRNA-142-3p Inhibits High-Glucose-Induced Endothelial-to-Mesenchymal Transition through Targeting TGF-Β1/Smad Pathway in Primary Human Aortic Endothelial Cells. Int. J. Clin. Exp. Pathol. 2018, 11, 1208–1217.
- Geng, H.; Guan, J. MiR-18a-5p Inhibits Endothelial-Mesenchymal Transition and Cardiac Fibrosis through the Notch2 Pathway. Biochem. Biophys. Res. Commun. 2017, 491, 329–336.
- Yuan, C.; Ni, L.; Yang, X.; Zhang, C.; Wu, X. Calcium-Sensing Receptor Participates in High Glucose-Induced EndMT in Primary Human Aortic Endothelial Cells. Front. Physiol. 2020, 11, 629542.
- Zhang, Y.; Dong, Y.; Xiong, Z.; Zhu, Z.; Gao, F.; Wang, T.; Man, W.; Sun, D.; Lin, J.; Li, T.; et al. Sirt6-Mediated Endothelial-to-Mesenchymal Transition Contributes Toward Diabetic Cardiomyopathy via the Notch1 Signaling Pathway. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 4801–4808.
- Du, J.-K.; Yu, Q.; Liu, Y.-J.; Du, S.-F.; Huang, L.-Y.; Xu, D.-H.; Ni, X.; Zhu, X.-Y. A Novel Role of Kallikrein-Related Peptidase 8 in the Pathogenesis of Diabetic Cardiac Fibrosis. Theranostics 2021, 11, 4207–4231.
- Peng, H.; Li, Y.; Wang, C.; Zhang, J.; Chen, Y.; Chen, W.; Cao, J.; Wang, Y.; Hu, Z.; Lou, T. ROCK1 Induces Endothelial-to-Mesenchymal Transition in Glomeruli to Aggravate Albuminuria in Diabetic Nephropathy. Sci. Rep. 2016, 6, 20304.
- Zhao, L.; Zhao, J.; Wang, X.; Chen, Z.; Peng, K.; Lu, X.; Meng, L.; Liu, G.; Guan, G.; Wang, F. Serum Response Factor Induces Endothelial-Mesenchymal Transition in Glomerular Endothelial Cells to Aggravate Proteinuria in Diabetic Nephropathy. Physiol. Genom. 2016, 48, 711–718.
- Yao, Y.; Li, Y.; Zeng, X.; Ye, Z.; Li, X.; Zhang, L. Losartan Alleviates Renal Fibrosis and Inhibits Endothelial-to-Mesenchymal Transition (EMT) Under High-Fat Diet-Induced Hyperglycemia. Front. Pharmacol. 2018, 9, 1213.
- Liu, W.; Wu, Y.; Yu, F.; Hu, W.; Fang, X.; Hao, W. The Implication of Numb-Induced Notch Signaling in Endothelial-Mesenchymal Transition of Diabetic Nephropathy. J. Diabetes Complicat. 2018, 32, 889–899.
- Chang, K.; Xie, Q.; Niu, J.; Gu, Y.; Zhao, Z.; Li, F.; Qin, Q.; Liu, X. Heparanase Promotes Endothelial-to-Mesenchymal Transition in Diabetic Glomerular Endothelial Cells through Mediating ERK Signaling. Cell Death Discov. 2022, 8, 67.
- Ma, Z.; Zhu, L.; Liu, Y.; Wang, Z.; Yang, Y.; Chen, L.; Lu, Q. Lovastatin Alleviates Endothelial-to-Mesenchymal Transition in Glomeruli via Suppression of Oxidative Stress and TGF-Β1 Signaling. Front. Pharmacol. 2017, 8, 473.
- Dong, R.; Zhang, X.; Liu, Y.; Zhao, T.; Sun, Z.; Liu, P.; Xiang, Q.; Xiong, J.; Du, X.; Yang, X.; et al. Rutin Alleviates EndMT by Restoring Autophagy through Inhibiting HDAC1 via PI3K/AKT/mTOR Pathway in Diabetic Kidney Disease. Phytomed. Int. J. Phytother. Phytopharm. 2023, 112, 154700.
- Guan, G.; Xie, J.; Dai, Y.; Han, H. TFPI2 Suppresses the Interaction of TGF-Β2 Pathway Regulators to Promote Endothelial-Mesenchymal Transition in Diabetic Nephropathy. J. Biol. Chem. 2022, 298, 101725.
- Liu, T.; Zhao, J.; Lin, C. Sprouty-Related Proteins with EVH1 Domain (SPRED2) Prevents High-Glucose Induced Endothelial-Mesenchymal Transition and Endothelial Injury by Suppressing MAPK Activation. Bioengineered 2022, 13, 13882–13892.
- Cao, X.; Song, Y.; Huang, L.-L.; Tian, Y.-J.; Wang, X.-L.; Hua, L.-Y. m6A Transferase METTL3 Regulates Endothelial-Mesenchymal Transition in Diabetic Retinopathy via lncRNA SNHG7/KHSRP/MKL1 Axis. Genomics 2022, 114, 110498.
- Cao, X.; Xue, L.-D.; Di, Y.; Li, T.; Tian, Y.-J.; Song, Y. MSC-Derived Exosomal lncRNA SNHG7 Suppresses Endothelial-Mesenchymal Transition and Tube Formation in Diabetic Retinopathy via miR-34a-5p/XBP1 Axis. Life Sci. 2021, 272, 119232.
- Wang, H.; Feng, Z.; Han, X.; Xing, Y.; Zhang, X. Downregulation of Acylglycerol Kinase Suppresses High-Glucose-Induced Endothelial-Mesenchymal Transition in Human Retinal Microvascular Endothelial Cells through Regulating the LPAR1/TGF-β/Notch Signaling Pathway. Can. J. Physiol. Pharmacol. 2022, 100, 142–150.
- Zhang, J.; Zeng, Y.; Chen, J.; Cai, D.; Chen, C.; Zhang, S.; Chen, Z. miR-29a/b Cluster Suppresses High Glucose-Induced Endothelial-Mesenchymal Transition in Human Retinal Microvascular Endothelial Cells by Targeting Notch2. Exp. Ther. Med. 2019, 17, 3108–3116.
- Cecoltan, S.; Ciortan, L.; Macarie, R.D.; Vadana, M.; Mihaila, A.C.; Tucureanu, M.; Vlad, M.-L.; Droc, I.; Gherghiceanu, M.; Simionescu, A.; et al. High Glucose Induced Changes in Human VEC Phenotype in a 3D Hydrogel Derived From Cell-Free Native Aortic Root. Front. Cardiovasc. Med. 2021, 8, 714573.
More