Diabetic Kidney Disease (DKD) is a significant complication of diabetes and primary cause of end-stage renal disease globally. The exact mechanisms underlying DKD remain poorly understood, but multiple factors, including the renin–angiotensin–aldosterone system (RAAS), play a key role in its progression. Aldosterone, a mineralocorticoid steroid hormone, is one of the key components of RAAS and a potential mediator of renal damage and inflammation in DKD. miRNAs, small noncoding RNA molecules, have attracted interest due to their regulatory roles in numerous biological processes. These processes include aldosterone signaling and mineralocorticoid receptor (MR) expression. Numerous miRNAs have been recognized as crucial regulators of aldosterone signaling and MR expression. These miRNAs affect different aspects of the RAAS pathway and subsequent molecular processes, which impact sodium balance, ion transport, and fibrosis regulation.
- Diabetic Kidney Disease
- microRNA
- aldosterone
- mineralocorticoid receptor
1. Introduction
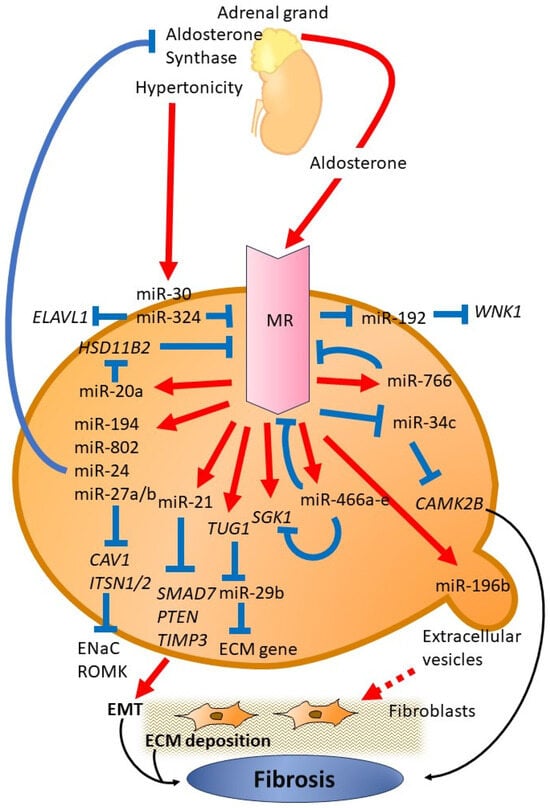
2. Function of miRNA
2.1. MiR-192
2.2. MiR-802
In studies involving rodent models, it was found that a high dietary intake of K+ stimulated the transcription and expression of miR-802 in the collecting ducts of the kidney. Consequently, this increase in miR-802 resulted in a decrease in caveolin-1 (CAV1) levels through a specific miR-802 seed sequence in the 3′ UTR of the CAV1 gene. As a consequence, the internalization of renal outer medullary potassium (ROMK) channels was reduced [24][34]. The regulation of CAV1 by miR-802 was confirmed through in vitro experiments, which demonstrated that a decrease in endogenous miR-802 led to an increase in CAV1 levels.2.3. MiR-194
The same research group conducted a study demonstrating that increased intake of dietary K+ also influences the regulation of ROMK channels through miR-194. Higher dietary K+ levels increase miR-194 expression, whereas low K+ levels decrease miR-194 levels. It was observed that the correlation between aldosterone release and miR-194 responded to dietary K+ levels. In addition, the researchers found that miR-194 reduces the expression of intersectin-1 (encoded by the ITSN1), which is a scaffold and regulatory protein. The reduction in intersectin-1 expression increases the presence of ROMK channels on the cell surface. This phenomenon is induced by delayed removal of ROMK channels from the plasma membrane, ultimately enhancing K+ transport [25][35].2.4. MiR-20a
11β-Hydroxysteroid dehydrogenase type 2 (11β-HSD2) is expressed in aldosterone-sensitive tissues and allows these tissues to respond to aldosterone through MR. Regulation of 11β-HSD2 expression is complex and involves CpG methylation, a number of transcription factors, and multiple microRNA binding sites. A reduction in 11β-HSD2 activity leads to MR activation, which results in sodium retention and salt-sensitive hypertension. To study the involvement of miRNA, two rat models with different 11β-HSD2 activity levels were compared: Sprague–Dawley rats with low activity and Wistar rats with high activity.2.5. MiR-23~24~27
The epithelial sodium channel (ENaC) plays a pivotal role in balancing Na+ levels and water in the kidney. Aldosterone primarily controls ENaC regulation in collecting duct epithelial cells. A recent study found that the levels of a group of miRNAs, mmu-miR-23–24–27, increase in response to aldosterone stimulation in the cortical collecting duct (CCD) of kidney nephrons [26][39]. Further investigations showed that miR-27a/b, members of this miRNA cluster, bind to the 3′ UTR of ITSN2 encoding intersectin-2, a protein in the distal kidney nephron that regulates membrane trafficking [26][39].Aldosterone induces miR-24 in MR-sensitive cells, including the epithelial cells of the distal nephron [26][39], and negatively regulates aldosterone production by targeting aldosterone synthase [27][40]. An analysis of healthy human adrenal tissue and aldosterone-producing adenoma showed distinct miRNA expression patterns, especially with significant differences in the expression of miR-24. Potential binding sites for miR-24 were found in the 3′ UTR of CYP11B1 (11β-hydroxylase) and CYP11B2 (aldosterone synthase) mRNAs. In vitro experiments confirmed that miR-24 can regulate CYP11B1 and CYP11B2 expression, thereby affecting cortisol and aldosterone [27][40].2.6. MiR-34c
Park et al. studied fibrotic signaling driven by the MR in the kidney and detected fibrosis-related proteins in mouse cortical duct cells (mCCD) upon aldosterone treatment [28][42]. They studied alterations in miRNA expression after aldosterone treatment and observed a number of changes, including in 15 miRNAs closely associated with the regulation of the Wnt signaling pathway. Further investigation revealed that miR-34c-5p specifically targeted Ca2+/calmodulin-dependent protein kinase type II beta-chain (CaMKIIβ). In aldosterone-treated cells, the reduction in miR-34c-5p levels increased CaMKIIβ mRNA and protein and stimulated fibronectin (FN) and alpha-smooth muscle actin. Transfection of mCCD with either CAMK2B siRNA or the miR-34c-5p mimic significantly reduced the FN induction by aldosterone, which was accompanied by decreased CaMKIIβ protein levels.2.7. MiR-196b
In db/db mice with diabetes, it was demonstrated that aldosterone increased proteinuria and the accumulation of tubulointerstitial extracellular matrix (ECM) [29][43]. However, the administration of MRA eplerenone alleviated the adverse effects of aldosterone. In coculture experiments, aldosterone-treated proximal tubular epithelial cells (PTECs) released extracellular vesicles (EVs). When these EVs were taken up by renal fibroblasts, an enhancement in ECM production was observed. Furthermore, injecting EVs from aldosterone-treated db/db mice into C57BL/6 mice resulted in increased ECM accumulation in the kidney. Subsequent miRNA analysis revealed that miR-196b-5p was the most elevated miRNA in EVs derived from PTECs after aldosterone stimulation. Further experiments revealed that overexpression of miR-196b-5p in fibroblasts increased ECM production, decreased suppressor of cytokine signaling 2 (SOCS2) expression, and resulted in elevated phosphorylation of the signal transducer and activator of transcription 3 (STAT3).2.8. MiR-766
A recent study demonstrated that miR-766 directly targets the MR gene NR3C2, thus promoting proliferation and metastasis in hepatocellular carcinoma [30][44]. The level of miR-766 correlates with the prognosis of liver cancer in such patients. In a different study, miR-766-3p served as a crucial regulator of inflammatory response in patients with rheumatoid arthritis (RA). Further analysis of plasma samples from patients with RA treated with abatacept revealed eight differentially expressed miRNAs, including miR-766-3p. MiR-766-3p indirectly reduced the activation of NF-κB; the same mechanism was involved in the reduction of MR expression. The anti-inflammatory effect of miR-766-3p was not limited to a specific cell type but suppressed inflammatory gene expression in various cell types, including human RA fibroblast-like synoviocytes (MH7A) and primary normal human mesangial cells (NHMCs) [31][45]2.9. MiR-466 Family
Another study investigated the increased expression of miRNA in mouse kidney after prolonged exposure to aldosterone for more than 3 days [32][46]. MiR-466a-e was found to be responsive to aldosterone, showing elevated levels both in vitro and in vivo in mCCD cells after aldosterone treatment. This miRNA family is known to target and regulate MR and SGK1 expression, consistent with the presence of target sites in the 3′ UTRs of these genes. Inhibition of these miRNAs significantly enhanced the expression of MR and SGK1, thus enhancing aldosterone sensitivity in mCCD.2.10. MiR-324-5p and MiR-30
Variations in extracellular tonicity across different nephron segments influence MR expression through mechanisms that are still not well understood [33][47]. Several RNA-binding proteins play a role in the posttranscriptional regulation of MR in response to extracellular tonicity. MiR-324-5p and miR-30c-2-3p, which are upregulated in renal cells under hypertonic conditions and in the kidneys of mice treated with furosemide, decrease MR expression [34][48]. These miRNAs directly affect the stability of MR transcripts by targeting NR3C2 and collaborate with tetradecanoyl phorbol acetate inducible sequence 11b (TIS11B) to degrade MR mRNA. In addition, these miRNAs inhibit ELAVL1 (HuR: Human antigen R) transcripts, consequently enhancing MR expression and signaling. The overexpression of miR-324-5p and miR-30c-2-3p modifies MR expression and signaling in KC3AC1 cells and attenuates aldosterone-regulated gene expression.2.11. Tug1-miR-29b
MiR-29b-3p has the potential to inhibit epithelial-to-mesenchymal transition (EMT) and fibrosis by regulating gene expression associated with the ECM in the kidney [35][36][51,52]. Cesana et al. [37][53] demonstrated a novel mechanism in which long noncoding RNAs (lncRNAs) control gene expression. LncRNAs serve as competing endogenous RNAs (ceRNAs), which bind to and sequester miRNAs, thereby inhibiting their interaction with target messenger RNAs (mRNAs). Bioinformatics analysis revealed that lncRNA Tug1 functions as a ceRNA and directly interacts with and sequesters miR-29b-3p. Furthermore, the study demonstrated that MR directly interacts with lncRNA Tug1, thus providing insight into the mechanisms of renal fibrosis involving MR and lncRNA signaling [38][54].2.12. MiR-21
MiR-21, which is one of the most extensively studied miRNAs, plays a significant role in various renal diseases, such as CKD, diabetic nephropathy, renal cell carcinoma, and renal fibrosis [39][55]. It is consistently upregulated in all these diseases [40][41][42][43][44][56,57,58,59,60]. Furthermore, miR-21 exacerbates renal fibrosis by enhancing profibrotic TGF-β signaling. It targets negative regulators of fibrosis, mothers against decapentaplegic homolog 7 (SMAD7), and phosphatase and tensin homolog (PTEN), thus leading to increased expression of ECM genes and other fibrotic factors [45][61].2.13. MiRNA-Based Therapeutic Strategies
Increased understanding of microRNAs in biology and their dysregulation in various diseases has prompted scientists to explore their potential for therapeutic applications. Two strategies have been employed. One strategy involves miRNA restoration therapy, where a downregulated or nonfunctional miRNA is supplied by a synthetic oligonucleotide, whereas the other strategy involves miRNA inhibition therapy, where miRNA overexpression is inhibited by antagonists [46][63]. MiRNA mimics used for therapeutic purposes are engineered to replicate the functions of endogenous miRNA. These synthetic double-stranded oligonucleotides undergo cellular processing to imitate the natural function of miRNA, thus offering enhanced stability and chemical modifications for efficient delivery and entry into target cells. Contrarily, inhibition of endogenous miRNA can be achieved by introducing anti-miRNA/ antagomiR oligonucleotides (AMOs). AMOs target pri-miRNA, pre-miRNA, or mature miRNA to either sequester or eliminate endogenous miRNAs [47][64]. There are various methods for delivering miRNAs, such as conjugation [48][67], virus-associated delivery [49][68], and nanoparticles [50][69]. Virus-associated miRNA delivery has shown experimental efficiency in cancers; however, safety concerns associated with viruses have limited its clinical application. Nonviral delivery systems appear more promising. The conjugation system, where lipids or cell receptor-targeting ligands are directly attached to miRNA, offers a potentially safer approach for miRNA delivery. While miRNA-based drugs are currently unavailable, numerous miRNA-based therapies for various human diseases are in clinical trials. These include Miravirsen (an LNA and phosphorothioate-modified antagomiR targeting miRNA-122), RG-101 (an N-acetyl-D-galactosamine-conjugated antagomiR targeting miR-122), RG-125 (an antagomiR targeting miR-103/107), RGLS5040 (an anti-miR-27), RG-012 (a phosphorothioate, 2′-O-methoxyethoxy-modified antagomiR targeting miR-21), MRG-201 (an LNA miRNA-29b mimic), MRX34 (an miRNA-34a mimic), MRG-106 (an LNA antagomiR targeting miR-155), MRG-107 (an antagomiR targeting miR-155), MRG-110 (an LNA antagomiR targeting miR-92), MesomiR (an miR-16 mimic), and ABX464 (a small molecular compound triggering miR-124 expression) [51][72].3. Summary
In summary, the prospect of using miRNAs to modulate aldosterone signaling and MR to mitigate DKD is indeed an exciting avenue of research. It offers the potential for more personalized, targeted, and highly effective treatments. However, the path forward still requires further research, comprehensive clinical trials, and collective efforts of the scientific community to fully unlock this therapeutic potential.