Human cytomegalovirus (HCMV) expresses a variety of viral regulatory proteins that undergo close interaction with host factors including viral-cellular multiprotein complexes. The HCMV protein kinase pUL97 represents a viral CDK ortholog (vCDK) that determines the efficiency of HCMV replication via phosphorylation of viral and cellular substrates. A hierarchy of functional importance of individual pUL97-mediated phosphorylation events has been discussed, however, the most pronounced pUL97-dependent phenotype could be assigned to viral nuclear egress, as illustrated by genetic ORF-UL97 deletion or pharmacological pUL97 inhibition. Despite earlier data pointing to a cyclin-independent functionality, experimental evidence increasingly emphasized the role of pUL97-cyclin complexes. Consequently, the knowledge about pUL97 involvement in host interaction, viral nuclear egress and additional replicative steps led to the postulation of pUL97 as an antiviral target. Indeed, validation experiments in vitro and in vivo confirmed the sustainability of this approach. Consequently, current investigations of pUL97 in antiviral treatment go beyond the known pUL97-mediated ganciclovir prodrug activation and henceforward include pUL97-specific kinase inhibitors. Among a number of interesting small molecules analyzed on experimental and preclinical stages, maribavir is presently investigated in clinical studies and, in the near future, might represent a first kinase inhibitor applied in the field of antiviral therapy.
- human cytomegalovirus (HCMV)
- protein kinase pUL97
- kinase-host interactions
- cyclin-dependent kinase (CDK) complexes
- regulatory mechanisms
- antiviral drugs
The Present Status of Controlling HCMV as a Major Human Pathogen
Molecular Biology of HCMV and Its Lytic Replication in Permissive Cells
HCMV, the prototypic β-herpesvirus, represents a major human pathogen and is characterized by a multifaceted mode of virus-host interaction. HCMV seroprevalence in the adult population ranges between approximately 40% to 90% and reaches even higher levels, of more than 95%, in countries with a low socio-economic standard [1]. HCMV exerts a strict species specificity and a comparably slow replication cycle spanning approximately three days in vitro [2][3]. Viral genomic DNA replication takes place in the nucleus and the double-stranded viral genome is packaged into capsids, which then undergo nuclear egress and budding through the nuclear membranes [4][5]. In the cytoplasmic virion assembly complex (cVAC), capsids are assembled with tegument proteins, before fully enveloped virus particles of approximately 150–200 nm are formed in the trans-Golgi network and released from the cell by final transition through the cytoplasmic membrane [2][6][7]. In addition to highly productive lytic infection of major target cells, such as fibroblasts, smooth muscle cells, endothelial and epithelial cells [8][9][10][11][12], HCMV causes life-long persistence by latent infection of minor target cells, such as monocytes/macrophages and CD34+ hematopoietic stem cells, in which latent HCMV may undergo reactivation resulting from immune insult, allogenic stimulation or differential signals (reviewed in [13]).
Pathogenesis of HCMV Infection
Due to the fact that primary and nonprimary infections (i.e., reactivation or reinfection) are mostly asymptomatic in healthy, immunocompetent individuals, HCMV infection usually remains clinically unrecognized. In contrast, patients with a compromised immune system, such as transplant recipients or AIDS patients, severely suffer from HCMV-related diseases, such as interstitial pneumonia, retinitis, gastroenteritis, esophagitis and organ failure, resulting in an increased mortality and morbidity [14][15][16][17]. Importantly, the immature immune system is a high risk factor for congenital cytomegalovirus infection (cCMV) of embryos and infants; thus, HCMV represents the most frequent cause for pathogen-derived developmental defects triggering mental retardation, loss of hearing or vision and microcephaly [18][19][20][21][22][23][24]. HCMV is one of few viruses that are able to cross the placenta efficiently, i.e., at least 33% of all primary infections during pregnancy of seronegative mothers, and an additional lower percentage of nonprimary infections undergo virus transmission resulting in cCMV infection of the unborn [25][26]. Thus, in Germany, approximately 3500 out of 700,000 newborns acquire cCMV per year [19]. Because of the lack of comprehensive HCMV screening, it is understood that approximately 10% of these are symptomatic at birth, including cases of stillbirth, and another 10%–15% may acquire symptoms at a later onset. HCMV can be transmitted by various body fluids, such as saliva, breast milk, vaginal secretions, semen and leukocytes containing blood and urine [27][28][29][30][31].
Current Options of Prevention and Control
Until today, no vaccine has been approved to control HCMV infections. Despite 60 years of intensive HCMV research, only a few antiviral drugs have been approved, which mostly interfere with the viral DNA polymerase pUL54, i.e., nucleoside/nucleotide analogs, such as the gold standard ganciclovir (GCV), its prodrug valganciclovir (VGCV), cidofovir (CDV) and the pyrophosphate analog foscarnet (FOS). Unfortunately, these drugs frequently cause severe side-effects, such as myelotoxicity, anemia and nephrotoxicity, or show poor bioavailability, which drives the selection of drug resistant virus variants [32][33][34][35][36][37]. In 2017, letermovir (LMV), the first anti-HCMV drug that targets the viral terminase complex consisting of pUL56, pUL89 and pUL51 core-subunits, was successfully assessed in clinical trials. Currently, LMV is approved for HCMV prophylaxis in hematopoietic stem cell transplantation recipients. LMV also represents a promising candidate for future combination therapies or even options of cCMV control [38][39][40][41]. However, based on the occurrence of LMV-resistant viral mutants [42] and the present lack of an approved treatment option for infants, the requirement of new antiviral drugs is still emphasized. This situation underlines the necessity of basic research to refine the understanding of the manifold and complex HCMV-host interplay and antiviral targeting strategies.
HCMV-Encoded Protein Kinase pUL97, a Multifunctional CDK Ortholog (vCDK)
Characteristics of the HCMV-Encoded Protein Kinase
pUL97 is a tegument protein, which is packaged into virions and is expressed with early-late kinetics [43]. The 707-amino acid protein exists in three isoforms due to alternative initiation of translation at residues M1, M74 or M157, resulting in protein varieties of approximately 100 kDa, 80 kDa and 70 kDa, respectively (Table 1, Figure 1) [44]. The full-length kinase possesses two NLS sequences in the poorly structured N-terminus, which mediate the predominantly nuclear localization of pUL97 [45][46]. The kinase domain was assigned to the globular C-terminal part, amino acids 337–651, based on sequence homologies or extended to 337–706, based on biochemical validation [47][48][49][50]. An invariant lysine residue at position 355 is essential for kinase activity, thus leading to the catalytically inactive K355M mutant [51][52][53]. Dimers and oligomers are formed via the self-interaction domain (amino acids 231–280) of pUL97 [54]. Interestingly, the direct association of pUL97 with human cyclins has been demonstrated and, hereby, the core region responsible for cyclin T1 binding proved to be identical with the pUL97 self-interaction domain [55], thus illustrating a functional role of cyclins in pUL97 dimerization/oligomerization [52][56][57][58]. Concerning the properties of protein interaction and substrate phosphorylation of pUL97, a number of viral as well as cellular proteins have been identified thus far [see references in legend of Figure 1]. The functionality of these substrates spans various regulatory aspects of viral replication, such as nuclear egress, intrinsic immunity, genome replication and gene expression (Table 1, Figure 1). Notably, several of the pUL97-specific substrate proteins also represent substrates of cellular CDK-cyclin complexes and may thus underlie a process of dual phosphorylation through these two different kinds of protein kinases in HCMV-infected cells. While sequence conservation between the open reading frame ORF-UL97 and other kinases is generally low, functional and structural similarities have been identified between pUL97 and CDKs, so that pUL97 was termed as a multifunctional viral CDK ortholog (vCDK). Importantly, both deletion of ORF-UL97 or pharmacological inhibition of pUL97 activity resulted in a strong delay of HCMV replication [52][59][60][61], likewise explained by the fact that the kinase exerts many regulatory functions during viral replication (Table 1). On this basis, pUL97 could be validated as an interesting target for novel antiviral strategies and a panel of small molecule-type inhibitors of pUL97 activity belonging to different chemical classes has been described during the last years (see below, Section 3, Section 4, Section 5 and Section 6).
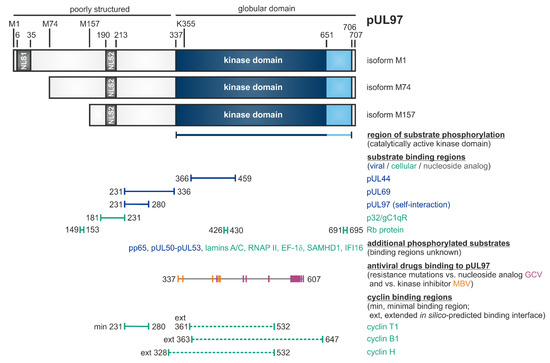
Figure 1. Schematic illustration of the modular structure and the so far identified binding regions within pUL97 [56]. The kinase domain is located between amino acids 337 and 706, as based on biochemical validation (or 337 and 651, as based on sequence homologies). K355 is an invariant lysine residue required for kinase activity. Expression of three pUL97 isoforms is determined by alternative translational initiation sites at M1, M74 and M157. Two nuclear localization signals (NLS1 and NLS2) are contained in the N-terminal unstructured portion of pUL97. Self-interaction/oligomerization of pUL97 is determined by amino acid region 231–280. This region overlaps with a minimal binding region for cyclin T1. Recent modeling approaches based on the in silico prediction of binding interfaces suggested extended binding interfaces for cyclins T1, B1 and H. Moreover, pUL97 is involved in the multiple regulatory steps during HCMV replication through the phosphorylation of viral and cellular substrates (see horizontal bars), as reported by several independent groups [44][45][46][54][55][57][62][63][64][65][66][67][68][69][70][71][72][73][74][75][76][77][78][79][80][81][82][83]. Substrates include the viral DNA polymerase cofactor pUL44, viral RNA transport factor pUL69, major tegument protein pp65, nuclear egress core protein heterodimer pUL50–pUL53, cellular multi-ligand binding protein p32/gC1qR, tumor suppressor/checkpoint protein Rb, nuclear lamins A/C, RNA polymerase II, translation factor EF-1δ, interferon-inducible proteins IFI16 and SAMHD1, as well as the therapeutically applied nucleoside analog ganciclovir (GCV; [47][56] and references therein). Interaction regions for GCV and the ATP-competitive pUL97 inhibitor maribavir (MBV) were defined by the location of resistance mutations detected so far (GCV: 405, 460, 466, 520, 590, 591, 592, 594, 595, 596, 597, 598, 599, 600, 601, 603, 607; MBV: 337, 353, 397, 409, 411). Note that this Figure represents a refined update, as adapted from an earlier version published elsewhere [56]; here, this also includes the hitherto mapped regions of resistance mutations against GCV and MBV, which possess high relevance for the discussion of an advanced antiviral drug targeting.
Table 1. Characterization of the molecular features and functional properties of the HCMV protein kinase pUL97.
| Property | General Description | Specific Feature | Own Findings (MM lab.) | Various References |
|---|---|---|---|---|
| Type of kinase | Ser/Thr | target site P + 5, target site LxSP |
[51] |
]. A central finding was that regions responsible for cyclin T1 interaction of pUL97 and pUL97-pUL97 self-interaction showed an overlap in N-terminal amino acids 231-280 (Figure 1; [54][134]). These data strongly suggest that cyclin binding is involved in pUL97-pUL97 self-interaction and very recent findings specified this activity for cyclin types T1 and H (but not B1), thus confirming the bridging function of cyclins T1/H in pUL97 dimerization or hetero-oligomerization. This self-interaction property is known to be a factor required for developing full catalytic activity of the pUL97 kinase [see references in Table 1]. The amino acid region 231–280 of pUL97 is considered as a minimal binding region for cyclin T1, which may be complemented by the additional binding of globular domain interfaces of pUL97 in the further C-terminal region, contributing to cyclin binding in a type-specific manner (cyclin T1, amino acids 361–532; cyclin B1, 363–647; cyclin H, 328–532; Figure 1; [55][56]).
In order to address the question of which spectrum of different types of human cyclins may associate with the viral pUL97 kinase, two specific experimental approaches have recently been performed. Firstly, a recombinant HCMV expressing a Flag-tagged version of pUL97 (namely the largest, fully functional isoform M1 of pUL97 encoded by HCMV AD169-UL97(Mx4)-Flag; [44]) was used for Flag-specific coimmunoprecipitation settings. The CoIP samples were then applied in a mass spectrometry-based (MS) proteomic assessment of pUL97-associated viral (Table S1) and cellular proteins (Table S2). HCMV AD169, expressing untagged pUL97, was used as a CoIP/MS specificity control. The identified viral proteins included several known interactors and/or substrates of pUL97 and showed a substantial overlap with those detected in our similar approach performed earlier, as based on the CoIP of pUL97-cyclin complexes using cyclin-specific antibodies [55]. Cellular proteins identified by this approach contained cyclins, CDKs and additional host proteins confirming earlier findings of pUL97-specific protein complexes. Notably, cyclins T1 and B1 were again safely detected, as those types of cyclins had been found by a variety of methodological approaches before (summarized in Table 2). Secondly, a panel of cyclin-specific antibodies were employed in a broader setting of CoIP analysis to learn more about the overall spectrum of pUL97-cyclin interaction. Representative members of the functional groups of cyclin types have been chosen, i.e., B-like, C-like and Y-like cyclins (Table 2, Supplementary Materials Figure 1). To this end, the cyclin-specific CoIP of pUL97 was then performed, again on the basis of total lysates prepared from HCMV-infected primary fibroblasts, followed by a quantitative assessment based on densitometric measurements (in duplicates, using two series of stained CoIP/Wb filters). The results, on the one hand, confirmed our earlier postulate that pUL97 strongly interacts with cyclin types B1, T1 and H (the latter primarily with pUL97 expressed in HCMV-infected cells, but very poorly with pUL97 transiently expressed in transfection-based settings; [55][56]). On the other hand, even more types of human cyclins could be additionally detected, either with moderate/weak (cyclins E, F and Y) or strong (cyclins B2 and K) properties of pUL97 interaction (Figure S1, Table S3 and Table 2). This topic of cyclin specificity of pUL97 and its functional relevance for HCMV replication will be further investigated by the use of recombinant HCMVs expressing mutant versions of pUL97 carrying cyclin-binding defects.
Table 2. Summarized findings of pUL97-cyclin interaction derived from complementary experimental settings *.
], major tegument protein pp65 [69], nuclear egress core proteins pUL50-pUL53 [73][78], cellular multiligand binding protein p32/gC1qR [72][76], tumor suppressor protein Rb [62], nuclear lamins A/C [57][67][68][72][80], RNAP II [74], translation factor EF-1δ [75][81][85], interferon-inducible, intrinsic immune restriction factors IFI16 [70] and SAMHD1 [115] (Figure 2; Table 3; compare with Table S1S3).
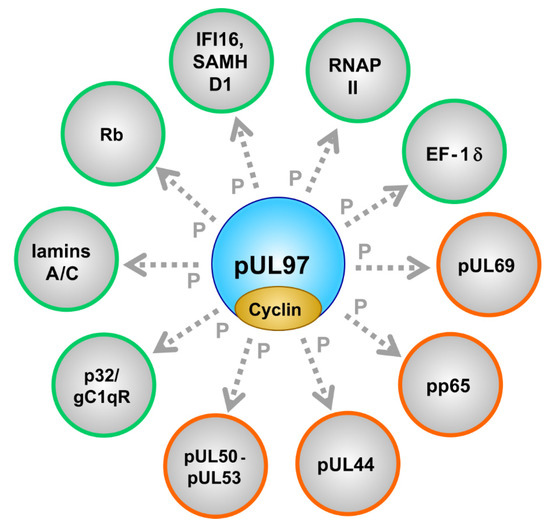
Figure 2. The cytomegalovirus-encoded CDK-like protein kinase pUL97 interacts with cyclins and phosphorylates a number of viral (encircled in orange) and cellular (encircled in green) substrate proteins.
Table 3. Characteristics of viral and cellular substrate proteins of the HCMV vCDK pUL97 as well as pUL97-associated cyclins.
| Protein Origin | Designation | Function | Remarks | References | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Viral | pUL50 | ] | [ | 87][88][89][90][91 | |||||||
| B-like | Cyclin A | + | ± | ] | |||||||
| - | - | . | . | core nuclear egress protein (NEC) | forms the NEC groove, multiple PPIs, phosphorylated by viral and cellular kinases | [62][77][78][110][ | Molecular mass, basic features | 100/80/70 kDa | isoforms due to alternative translational start sites | [44][45][54] | [ |
62][147][148] (Table 4). In addition, the suppression of CDKs 1, 2, 5 and 9 by indirubin-derivatives increases the HCMV-inhibitory effect of maribavir (MBV), a potent pUL97 inhibitor [58]. Thus, pUL97 and CDKs possess at least partially overlapping functions.
Table 4. Comparison of distinct molecular characteristics shared between vCDK pUL97 and human CDKs.
| Kinase Characteristics | pUL97 | CDK1 | CDK7 | CDK9 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 136 | ] | [ | 137 | ] | |||||||||||||
| 43 | ] | [ | 52 | ][92] | |||||||||||||
| . | . | ||||||||||||||||
| Amino acids (aa) | 707 | 297 | 345 | 372 | |||||||||||||
| Cyclin B1 | + | + | + | Viral | pUL53 | ||||||||||||
| Aa sequence identity to pUL97 | 100% | - | core nuclear egress protein (NEC) | forms NEC hook, possibly docking to capsids, phosphorylated by viral kinase | [ | 4.5%73 | 4.2%][138][139] | + | + | . | + | Expression pattern | three isoforms M1, M74, M15 (referring to other herpesviral protein isoforms) | differences in substrate binding, nuclear translocation and drug susceptibility | [ | ||
| Cyclin B2 | - | 44 | + | ][93] | [94][95][96 | ||||||||||||
| 8.6% | ] | [ | Viral | ||||||||||||||
| Cyclin binding partner [56 | pUL44 | ]DNA polymerase pUL54 processivity factor | 97 | ] | |||||||||||||
| phosphorylation might regulate activity | [149][150] | cyclin B1 cyclin H | - | - | - | cyclin T1[ | cyclin A1/A2 cyclin B1/B2/B3 cyclin D1/D3 cyclin F cyclin K | . | . | . | (activating) | cyclin H cyclin A2 cyclin B1/B2 cyclin E (activating)76][114] |
|||||
| cyclin T1/T2 | cyclin H | cyclin K (activating) |
Similarity and sequence conservation with other kinases | low | <35% identity with herpesviral kinases, <15% identity with cellular kinases | [45][85] | [ | Viral48 | pp65] | major tegument protein[49] | |||||||
| Cyclin D1 | - | [ | - | 50] | - | [62] | |||||||||||
| - | - | - | . | . | massively phosphorylated and virion-associated with pUL97 | ||||||||||||
| Region in the kinase required for cyclin binding [55][151][152] | [ | cyclin T1: 231ESQDSAVASGPGRIPQPLSGSSGEESATAVEADSTSHDDVHCTCSNDQII280 and in silico-predicted binding interfaces for cyclins B1, H and T1 spanning aa 328–647 | 44 | ][60][69] | |||||||||||||
| cyclin B1: a positively charged region in the N-lobe (containing K6, K9, K34, R36, R75, excluding the PSTAIRE helix) | cyclin A2: 45PSTAIRE | Sequence conservation ORF-UL97 of HCMVs | high | no variation of translational start sites, NLS sequences or kinase domains | [44] | [ | |||||||||||
| Cyclin E | ± | 98 | ± | ][ | - | 99] | |||||||||||
| - | . | . | . | . | Viral | [pUL69 | 149][153][154RNA transport regulator | ][155][156]phosphorylation regulates activity | cyclin B1 | cyclin B1 S126 by CDK1 S128 by CDK1 | cyclin H by CDK7/CDK8-cyclin C (inhibitory)[63][100] | n.d.*[140][141 | Related to cell kinases | cyclin-dependent kinases (CDKs), viral CDK ortholog | functional overlap with CDKs, specific crosstalk with CDK9, CDK7 and CDK1, direct interaction with cyclins | [47][55][56][100][63][64][65][66] | [ |
| ] | Cyclin F | . | 57 | ±][ | |||||||||||||
| 67 | ] | Viral | [ | 101] | |||||||||||||
| - | - | . | . | . | . | ||||||||||||
| T-loop phosphorylation [56][ | pUL97 | CDK-like serine/threonine protein kinase, multifunctional | dimers/oligomers, autophosphorylation | 157][158][159][160][161][162][163][164] | no, (possibly S483) | T161 by CAK (activating) | S164 and T170 by CDK1/CDK2 (activating)[50][53][54][83][87][120][124] | Coregulation of viral replication by pUL97 and cellular kinases | several novel cellular kinases, including CDKs, identified to be involved in HCMV replication | virus-supporting functions in signaling pathways and nuclear capsid egress | [55][56][102][103] | ||||||
| C-like | Cyclin H | [ | + | 104][105][106 | + | ][107][108][109] | |||||||||||
| T186 by CaMK1D or CDK9 (S175 by CAK, not essential for activity) | Cellular | p32/gC1qR | |||||||||||||||
| Autophosphorylation | multiligand binding protein, multifunctional | [120] | - | [158]NEC bridging factor | [159] | yes | no[68][72 | - | ][ | + | 142 | - | - | ] | - | Substrate proteins | viral, cellular |
| (yes) outside the T-loop | yes within the T-loop | pUL44, pUL69, pp65, Rb, p32/gC1qR, nuclear lamins, EF-1δ, RNAP II, IFI16, SAMHD1 | [53][63][103][68][69][ | ||||||||||||||
| Cyclin K | Cellular | ||||||||||||||||
| Rb phosphorylation [55][62][88][ | 70 | ] | [71][72 | . | ] (references therein) | 148][lamins A/C[ | 165]structural and regulatory components of the nuclear envelope57][62][67][ | [lamin phosphorylation is a rate-limiting step of viral nuclear egress110][74][75][111][112][113][114][115][116 | + | ] (see also refs. in Figure 1) | |||||||
| - | - | . | . | . | . | 166 | Involvement in intrinsic immunity evasion | stimulation of viral counterdefense of immunity | interaction with cellular restriction factors IFI16 and SAMHD1 | [70][ | |||||||
| Cyclin L2a | 117 | ] | [ | 118] | |||||||||||||
| . | - | - | - | . | . | . | . | Auto-phosphorylation | pronounced auto-phosphorylation activity, several N-terminal Ser and Thr residues | autophosphorylation most probably required for kinase activity/autoactivation | [44][54][56][68][119] | [87][88][120] | |||||
| Cyclin T1 | + | + | + | + | + | + | + | - | Nucleoside phosphorylation | ganciclovir, valganciclovir, penciclovir, acyclovir, etc. | prodrug-activating monophosphorylation as an essential step in antiviral therapy | [51][121] | [59][122][123] | ||||
| Y-like | Cyclin Y | [ | . | 124] | ±[125] | ||||||||||||
| - | - | . | . | . | . | Incorporation into virions | component of virion tegument | virion-derived pUL97 possesses highly detectable kinase activity | [45][69] | [43][126][127] | |||||||
| Intracellular localization | mainly nuclear | two nuclear localization signals, NLS-1 (6–35), NLS-2 (164–213), classical importin-α pathway | [45][46][71][85][128] | [60] | |||||||||||||
| Inhibitors of pUL97 | small molecules (<500 Da, various chemical classes) | indolocarbazoles, benzimidazoles, quinazolines, others | [53] (references therein) [44][86][129] | [124][130][131] | |||||||||||||
| Phenotype of pUL97 inhibition or UL97 deletion | strongly reduced viral replication efficiency (100–1000-fold) | delayed replication kinetics; impaired genomic replication; impaired viral nuclear egress | [44][51][53][68][119][76][132] | [59][60][114][133] |
The interaction between HCMV pUL97 and human cyclins of the types B1, T1 and H has been described in our earlier reports [47][55][134]. The three cyclins obviously possess different affinities in terms of strength of pUL97 binding detected by coimmunoprecipitation (CoIP)- and mass spectrometry (MS)-based analyses. In case of cyclin B1, a requirement of catalytic activity of pUL97 for cyclin binding was identified, whereas in case of cyclin H, pUL97 interaction was found dependent on the environment of HCMV replication [55]. Recently published data indicate a substrate-bridging function of cyclin(s) for the binding of pUL97 to its substrate pp65, as determined with a pp65 mutant lacking a putative cyclin-docking motif [66].
Previous investigations led to the postulate of a substantial relevance of pUL97-cyclin interactions, as characterized by the following findings: (i) The HCMV kinase pUL97 acts as a structural CDK ortholog originally based on our bioinformatic modeling and biochemical analyses. (ii) Our initial report on pUL97-cyclin T1 interaction could be extended to additional types such as cyclins B1 and H [47][55][56][134]. (iii) The interaction pUL97-cyclins B1/T1/H was confirmed by several methods including highly sensitive mass spectrometry-based proteomics. (iv) Specifically, the interaction pUL97-cyclin B1 was found to be phosphorylation-dependent for both proteins. In addition, cyclin B1 (but not H) was phosphorylated by pUL97 in vitro [56]. (v) Using a protein assembly-based CoIP assay, the formation of binary and ternary complexes involving pUL97, cyclin H and CDK7 was identified, thus suggesting a cyclin bridging concept [135
*Data on pUL97-cyclin interaction were derived from the experimental settings of either mass 188 spectrometry-based proteomics (MS) or Western blot detection (Wb), both performed by the use of 189 coimmunoprecipitates derived from cyclin-specific immunoprecipitation (cyclin IP) or pUL97 190 immunoprecipitation (pUL97 IP). Colocalization patterns between pUL97 and individual cyclins, in 191 particular nuclear punctate patterns of accumulation in viral replication centers, were determined by 192 indirect immunofluorescence (IF) double-stainings and confocal imaging. Recombinant expression of 193 pUL97 and/or cyclins was performed by transient transfected of 293T cells (transfection), yeast cells 194 (yeast two-hybrid assay) or bacterial expression systems, the latter for analyzing the phosphorylation 195 of recombinant cyclins by transfection-derived pUL97 in the in vitro kinase assay (IVKA). In panel A, 196 the criteria of categorization were set as follows: +, strong pUL97-cyclin interaction (MS: WSC ≥4; Wb: 197 % IP values > 20% IP control and ≥15-fold above Flag neg. control); ±, weak interaction (MS: WSC = 3; 198 Wb: % IP values >20% IP control or ≥15-fold above Flag neg. control); -, no detectable interaction; ., 199 not determined.
Phosphorylation of a Panel of Regulatory Viral Proteins and Host Factors through pUL97
| ] | |||||||
| [ | |||||||
| 57 | |||||||
| ] | |||||||
| [ | |||||||
| 67 | |||||||
| ] | |||||||
| [ | |||||||
| 68 | ] | [ | 71 | ][ | S780, S807, S811, T821, T823, T82680][143] | ||
| S249, T252, T373, S807, S811 | no | C-terminus (793–834) | Cellular | ||||
| p53 phosphorylation [167][168][ | Rb | retinoblastoma protein, cell cycle check-point regulator | multiply phosphorylated by CDKs and pUL97 | 169][48 | n.d.][ | S31557][101][113][116] | |
| S33 (MAT1-dependent) | S33, S315, S392 | Cellular | IFI16 and SAMHD1 | intrinsic immune restriction factors of virus infections | interferon-induced, phosphorylation-controlled | ||
| Lamin A/C phosphorylation [67][143][170][171] | [ | 70 | ] | [115][117 | S22 (inhibitory)][118 | S22, S392 (inhibitory)][ | no144] |
| no | Cellular | ||||||
| CTD RNAP II phosphorylation [74][ | RNAP II | 172][173] | S2, S5 (activating)main cellular mRNA transcriptase | activity-regulated by C-terminal phosphorylation (CTD) | [59 | no][61][74 | S2, S5, S7 (activating)][106] |
| S2, S5, S7 (activating) | Cellular | EF-1 | translation elongation factor 1 delta | activity-regulated by phosphorylation | [53][75] | ||
| Cellular | cyclins | regulatory subunits of CDKs | types B1, H, T1 were found pUL97-associated (possibly also B2, K, others) | [55][56][58][67][108] |
It should be emphasized that the pUL97 substrate proteins belong to several functionally different groups (Table 3), thus underlining the multifunctional nature of this singly expressed viral protein kinase. Viral proteins interacting with and being phosphorylated by pUL97 span the regulatory areas of viral nuclear egress (pUL50-pUL53 core NEC), genome replication (pUL44), tegumentation and immune-regulatory functions (pp65), viral RNA transport (pUL69) and the pUL97-pUL97 autophosphorylation/autoregulation associated with the formation of dimers and oligomers. As far as cellular substrates are concerned, the following regulatory areas are addressed: nuclear egress (lamins A/C, p32/gC1qR), cell cycle control (Rb, cyclins), intrinsic immune regulation (IFI16, SAMHD1) and transcription/translation (RNAP II, EF-1δ). The entity of this spectrum of pUL97-driven processes in virus-infected cells illustrates the functional importance of pUL97 for a high efficiency of viral replication, as demonstrated by the defects of recombinant viruses carrying UL97 deletions/mutations (up to factor 100–1000). Interestingly, the dimension of a replication defect resulting from drug-inhibited pUL97 was demonstrated to be more drastic in non-cycling compared to cycling cells [145], probably referring to the crosstalk and functional complementation between active cellular CDK-cyclin complexes and the vCDK. Moreover, the complex patterns of protein-protein interactions (PPI) undergone by pUL97 have recently been revealed by the use of highly sensitive mass spectrometry-based proteomic and phosphoproteomic approaches [55][56][72][88]. These findings make the occurrence of higher-order, pUL97-associated PPI complexes seem highly likely.
HCMV pUL97 and Related Herpesviral vCDKs
Most pUL97-related herpesviral kinases function as viral CDK orthologs (vCDKs). They were also termed conserved herpesviral protein kinases (CHPKs), as encoded by a gene conserved throughout the family Herpesviridae (e.g., prototype pUL97 and homologous kinases). Despite conservation of the UL97 gene locus, substantial variation of the primary coding sequence has been identified between herpesviruses. In addition to CHPKs, a second protein kinase is encoded by an additional non-conserved gene restricted to the subfamily α-Herpesvirinae (e.g., prototype pUS3 kinase of herpes simplex virus). CDK activity has been shown to be involved in multiple steps during HCMV infection [146]. vCDKs phosphorylate typical CDK substrates such as Rb and lamins A/C and show CDK activity in a yeast complementation assay [57][62][67][80]. The Saccharomyces cerevisiae mutant lacking activity of its sole CDK, cdc28, shows growth arrest in the early S/late G1 phase, which is overcome by CDK1 (human), pUL97 (HCMV), pU69 (HHV-6 and -7) and BGLF4 (EBV) expression [57]. In addition, pUL97 and CDK share substrate proteins, such as pUL69, RNAP II and EF-1δ [63][75][81][100]. Of note, pUL97 and CDKs phosphorylate Rb at the same residues (S780, S807, T821), leading to the inactivation of the cell cycle-inhibitory and tumor suppressor functions of Rb [
Validation of vCDK pUL97 as an Antiviral Target and Various pUL97 Inhibitors Explored as Experimental Antiviral Drugs
Role of the pUL97 Kinase in Anti-HCMV Standard Therapy
The HCMV-encoded CDK ortholog pUL97 has significance in the therapy of HCMV infections, as it is responsible for the phosphorylation-mediated activation of GCV/VGCV, still representing the therapy gold standard and, similarly, additional nucleosides such as acyclovir (ACV), penciclovir (PCV) and others [91][123][177]. Hereby, the specific role of pUL97 is that nucleoside analogs have to be initially monophosphorylated in a step catalyzed by pUL97 kinase [90]. Thereafter, the active triphosphate metabolites have to be generated in a series of steps of further phosphorylation catalyzed by human guanylate kinase, dGMP kinase, phosphoglycerate kinase and potentially other host kinases [25]. In the triphosphate form, these analogs represent the active antiviral determinants, then acting as a substrate of the HCMV DNA polymerase, ultimately inhibiting the elongation of viral genome synthesis.
Target Validation and pUL97 Inhibitors
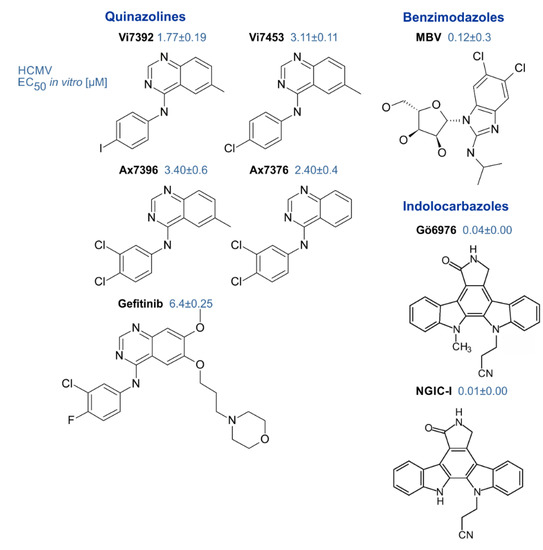
Genetic mutation studies showed that pUL97 plays a rate-limiting regulatory role for the replication efficiency of HCMV and virus titers were reduced by orders of magnitude when the coding sequence was disrupted [59][61]. Moreover, pharmacological inhibition of pUL97 activity by small molecules derived from various chemical classes blocked viral replication in a manner corresponding to the pUL97 null phenotype and thus proved to be a potent antiviral targeting strategy [178]. Since then, the pharmacologic inhibition of pUL97 activity together with genetic techniques have helped to characterize the mechanisms of pUL97 supporting the viral replication and virus–host kinase interactions [52][53][56][179]. A number of inhibitors of pUL97 kinase activity have been identified that exert potent antiviral activity against HCMV [86][129][178][180]. These include indolocarbazoles [51][119][130], quinazolines [86][132][181] and benzimidazole analogs [178] (Figure 3). A number of detailed investigations, both on cell culture-based in vitro and preclinical in vivo animal models, underlined the high value of this antiviral approach (reviewed in [25][53][83][182]). Thus far, however, with the exception of maribavir, none of these compounds has progressed to clinical studies.
Clinical Investigation of the First Prototype of a Kinase Inhibitor in Antiviral Treatment: Maribavir
MBV is a benzimidazole riboside, structurally related to the terminase inhibitors BDCRB and GW275175X [25]. This molecule exerts outstanding inhibitory activity against the pUL97 kinase and shows very low levels of side/off-target effects [183]. MBV exhibits favorable pharmacokinetic properties, is well tolerated and holds promise as a new drug for the treatment of HCMV infections [184][185][186]. Thus, MBV represents a novel developmental drug that might become the first prototype of a kinase inhibitor in antiviral treatment. In the first phase III clinical study, maribavir-treated patients failed to meet the clinical endpoint objectives [187]. Currently further phase III trials are enrolling patients to compare the efficacy of MBV with GCV, and this clinical development is currently continuing (NCT02931539, NCT02927067). One limitation might be based on the fact that the inhibition of pUL97 kinase activity by MBV interferes with the activation of GCV, thus resulting in drug antagonism, which most probably reduces their antiviral efficacies in a combination therapy [133][188][189]. Mutations conferring MBV resistance are distinct from those conferring GCV resistance, with sites of mutations partly located outside the conserved kinase domains [83][124][190]. In rare cases, kinase domain mutations arise in the laboratory that are essentially kinase null mutations and can confer resistance to MBV or GCV [182]. Notably, however, MBV exerts activity against typical GCV-resistant strains and might therefore create new options in the treatment of drug-resistant HCMV infections [178][191][192]. Interestingly, the three different isoforms of the kinase also show altered susceptibility of the virus to MBV [44]. An additional type of an intermediate-level MBV-resistance has been identified for viral variants carrying mutations, not in the UL97 but rather in the UL27 gene [193][194]. To date, it is not clear whether resistance mutations in UL27 would arise in clinical settings, since in animals the deletion of ORF-UL27 resulted in a modest half-log reduction in viral in vitro replication capacity, with no apparent effect on replication in vivo [195].
The Relevance of Targeting a Herpesviral Kinase Activity in Antiviral Strategies
The HCMV-encoded kinase pUL97 combines two different aspects of medical importance, namely serving as promoter of prodrug activation through the activating monophosphorylation of GCV, VGCV and related nucleoside analogs and as a validated target of antiviral kinase inhibitors. The currently ongoing clinical investigations of MBV are approaching an exciting interim phase and it will be highly relevant to see whether this drug candidate achieves primary endpoints. MBV might not only represent a novel drug for the treatment and prevention of HCMV disease but it would likewise be a very promising novel prototype of a kinase inhibitor that might—compared to the numerous currently approved kinase inhibitors in antitumoral treatments—for the first time enter the field of antiviral therapy. Notably, the applicability of a further mode of action of antiviral drugs would directly broaden the options of overcoming previous problems with antiviral drug resistance. The pharmacological interference with viral kinase activity/protein phosphorylation by MBV, in addition to the targeting of viral genome replication/polymerase activity (GCV) and viral terminase activity/genome processing (LMV), would open a third mechanistic option of HCMV treatment. Thus, resistant mutants arising from GCV and LMV treatment would very probably remain susceptible to MBV treatment, so that variable regimens might become available, possibly including combination therapies. It should be mentioned, however, that GCV and MBV combination would underlie an antagonistic principle, due to the two counteractive roles of pUL97 in such a case (prodrug converting GCV phosphorylation through active pUL97 versus an inhibition of pUL97 activity by MBV). Nevertheless, other combinations between MBV and LMV, GCV and LMV or the involvement of additional approved anti-herpesviral drugs, such as CDV, ACV etc., might lead to a substantial improvement of medication regimens. In this sense, anti-HCMV therapy might also greatly benefit from the experiences made in the field of human immunodeficiency virus/AIDS during the past decades, as mostly gathered by the steady development of novel antiretroviral combination therapies.
References
- Raskit Lachmann; Anna Loenenbach; Tim Waterboer; Nicole Brenner; Michael Pawlita; Angelika Michel; Michael Thamm; Christina Poethko-Müller; Ole Wichmann; Miriam Wiese-Posselt; Cytomegalovirus (CMV) seroprevalence in the adult population of Germany. PLOS ONE 2018, 13, e0200267, 10.1371/journal.pone.0200267.
- Mocarski, E.S.; Shenk, T.; Griffiths, P.D.; Pass, R.F. Cytomegaloviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 1960–2014.
- Roizman, B.; Knipe, D.M. Herpesviruses and Their Replication; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; pp. 2399–2460.
- Stinski, M.F. Molecular Biology of Cytomegaloviruses. In The Herpesviruses; Viruses, B.R., Ed.; Springer: Boston, MA, USA, 1983; pp. 67–113.
- Chung-Pei Lee; Mei-Ru Chen; Escape of herpesviruses from the nucleus. Reviews in Medical Virology 2010, 20, 214-230, 10.1002/rmv.643.
- Subhendu Das; Amit Vasanji; Philip E. Pellett; Three-Dimensional Structure of the Human Cytomegalovirus Cytoplasmic Virion Assembly Complex Includes a Reoriented Secretory Apparatus▿ †. Journal of Virology 2007, 81, 11861-11869, 10.1128/JVI.01077-07.
- Veronica Sanchez; Kenneth D. Greis; Elizabeth Sztul; William J. Britt; Accumulation of Virion Tegument and Envelope Proteins in a Stable Cytoplasmic Compartment during Human Cytomegalovirus Replication: Characterization of a Potential Site of Virus Assembly. Journal of Virology 2000, 74, 975-986, 10.1128/jvi.74.2.975-986.2000.
- Bodo Plachter; Christian Sinzger; Gerhard Jahn; Cell Types Involved in Replication and Distribution of Human Cytomegalovirus. Advances in Applied Microbiology 1996, 46, 195-261, 10.1016/s0065-3527(08)60073-1.
- Sinzger, C.; Digel, M.; Jahn, G; Cytomegalovirus cell tropism. Curr. Top. Microbiol. Immunol. 2008, 325, 63–83.
- Christian Sinzger; Annemarie Grefte; Bodo Plachter; Annette S. H. Gouw; T. Hauw The; Gerhard Jahn; Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. Journal of General Virology 1995, 76, 741-750, 10.1099/0022-1317-76-4-741.
- Weng, C.; Lee, D.; Gelbmann, C.B.; Van Sciver, N.; Nawandar, D.M.; Kenney, S.C.; Kalejta, R.F; Human Cytomegalovirus Productively Replicates In Vitro in Undifferentiated Oral Epithelial Cells. Journal of Virology 2018, 92, e00903-18, 10.1128/JVI.00903-18.
- Scrivano, L.; Sinzger, C.; Nitschko, H.; Koszinowski, U.H.; Adler, B; HCMV Spread and Cell Tropism are Determined by Distinct Virus Populations. PLOS Pathogens 2011, 7, e1001256, 10.1371/journal.ppat.1001256.
- Donna Collins-McMillen; Jason C. Buehler; Megan Peppenelli; Felicia Goodrum; Molecular Determinants and the Regulation of Human Cytomegalovirus Latency and Reactivation. Viruses 2018, 10, 44.
- M Boeckh; W Garrett Nichols; Genovefa A. Papanicolaou; Robert Rubin; John R Wingard; John Zaia; Cytomegalovirus in hematopoietic stem cell transplant recipients: current status, known challenges, and future strategies. Biology of Blood and Marrow Transplantation 2003, 9, 543-558, 10.1016/s1083-8791(03)00287-8.
- Rafailidis, P.I.; Mourtzoukou, E.G.; Varbobitis, I.C.; Falagas, M.E; Severe cytomegalovirus infection in apparently immunocompetent patients: a systematic review. Virology Journal 2008, 5, 47.
- C. Steininger; Clinical relevance of cytomegalovirus infection in patients with disorders of the immune system. Clinical Microbiology and Infection 2007, 13, 953-963, 10.1111/j.1469-0691.2007.01781.x.
- Dana G. Wolf; Nell S. Lurain; Tsila Zuckerman; Ron Hoffman; Judith Satinger; AliK Honigman; Niveen Saleh; Emanuel S. Robert; Jacob M. Rowe; Zipora Kra-Oz; Emergence of late cytomegalovirus central nervous system disease in hematopoietic stem cell transplant recipients. Blood 2003, 101, 463-465, 10.1182/blood-2002-07-1982.
- William J. Britt; Congenital Human Cytomegalovirus Infection and the Enigma of Maternal Immunity. Journal of Virology 2017, 91, e02392-16, 10.1128/JVI.02392-16.
- Horst Buxmann; Klaus Hamprecht; Matthias Meyer-Wittkopf; Klaus Friese; Primary Human Cytomegalovirus (HCMV) Infection in Pregnancy. Deutsches Aerzteblatt Online 2017, 114, 45-52, 10.3238/arztebl.2017.0045.
- Tania Crough; Rajiv Khanna; Immunobiology of Human Cytomegalovirus: from Bench to Bedside. Clinical Microbiology Reviews 2009, 22, 76-98, 10.1128/cmr.00034-08.
- Stuart T. Hamilton; Wendy Van Zuylen; Antonia Shand; Gillian M. Scott; Zin Naing; Beverley Hall; Maria E Craig; Bill Rawlinson; Prevention of congenital cytomegalovirus complications by maternal and neonatal treatments: a systematic review. Reviews in Medical Virology 2014, 24, 420-433, 10.1002/rmv.1814.
- M. G. Revello; Giuseppe Gerna; Human cytomegalovirus tropism for endothelial/epithelial cells: scientific background and clinical implications. Reviews in Medical Virology 2010, 20, 136-155, 10.1002/rmv.645.
- Sia, I.G.; Patel, R; New Strategies for Prevention and Therapy of Cytomegalovirus Infection and Disease in Solid-Organ Transplant Recipients. Clin. Microbiol. Rev. 2000, 13, 83–121.
- Yoshihiro Tsutsui; Effects of cytomegalovirus infection on embryogenesis and brain development. Congenital Anomalies 2009, 49, 47-55, 10.1111/j.1741-4520.2009.00222.x.
- Britt, W.J.; Prichard, M.N; New therapies for human cytomegalovirus infections. Antivir. Res. 2018, 159, 153–174.
- Aileen Kenneson; Michael Cannon; Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Reviews in Medical Virology 2007, 17, 253-276, 10.1002/rmv.535.
- T. H. Weller; J. C. Macauley; J. M. Craig; P. Wirth; Isolation of Intranuclear Inclusion Producing Agents from Infants with Illnesses Resembling Cytomegalic Inclusion Disease. Experimental Biology and Medicine 1957, 94, 4-12, 10.3181/00379727-94-22841.
- Harris D. Riley; History of the Cytomegalovirus. Southern Medical Journal 1997, 90, 184-190, 10.1097/00007611-199702000-00004.
- Rowe, W.P.; Hartley, J.W.; Cramblett, H.G; Detection of human salivary gland virus in the mouth and urine of children.. American journal of hygiene 1958, 67, 57–65.
- P. R. Wallace; W. H. Janet; W. Samuel; C. T. Horace; J. H. Robert; Cytopathogenic Agent Resembling Human Salivary Gland Virus Recovered from Tissue Cultures of Human Adenoids. Experimental Biology and Medicine 1956, 92, 418-424, 10.3181/00379727-92-22497.
- M. G. Smith; Propagation in Tissue Cultures of a Cytopathogenic Virus from Human Salivary Gland Virus (SGV) Disease. Experimental Biology and Medicine 1956, 92, 424-430, 10.3181/00379727-92-22498.
- Biron, K.K; Antiviral drugs for cytomegalovirus diseases. Antivir. Res. 2006, 71, 154–163.
- Guy Boivin; N. Goyette; C. Gilbert; A. Humar; E. Covington; Clinical impact of ganciclovir-resistant cytomegalovirus infections in solid organ transplant patients. Transplant Infectious Disease 2005, 7, 166-170, 10.1111/j.1399-3062.2005.00112.x.
- Lara Danziger-Isakov; G. Mark Baillie; Hematologic complications of anti-CMV therapy in solid organ transplant recipients. Clinical Transplantation 2009, 23, 295-304, 10.1111/j.1399-0012.2008.00942.x.
- Georg Härter; Detlef Michel; Antiviral treatment of cytomegalovirus infection: an update. Expert Opinion on Pharmacotherapy 2012, 13, 623-627, 10.1517/14656566.2012.658775.
- Peter Lischka; H. Zimmermann; Antiviral strategies to combat cytomegalovirus infections in transplant recipients. Current Opinion in Pharmacology 2008, 8, 541-548, 10.1016/j.coph.2008.07.002.
- Einat Shmueli; Reuven Or; Michael Y. Shapira; Igor B. Resnick; Orit Caplan; Tali Bdolah-Abram; Dana G. Wolf; High Rate of Cytomegalovirus Drug Resistance Among Patients Receiving Preemptive Antiviral Treatment After Haploidentical Stem Cell Transplantation. The Journal of Infectious Diseases 2013, 209, 557-561, 10.1093/infdis/jit475.
- Chong, P.P.; Teiber, D.; Prokesch, B.C.; Arasaratnam, R.J.; Peltz, M.; Drazner, M.H.; Garg, S. Letermovir successfully used for secondary prophylaxis in a heart transplant recipient with ganciclovir-resistant cytomegalovirus syndrome (UL97 mutation). Transplant. Infect. Dis. 2018, 20, e12965.
- Goldner, T.; Hewlett, G.; Ettischer, N.; Ruebsamen-Schaeff, H.; Zimmermann, H.; Lischka, P; The novel anticytomegalovirus compound AIC246 (Letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J. Virol. 2011, 85, 10884-10893.
- Lischka, P.; Hewlett, G.; Wunberg, T.; Baumeister, J.; Paulsen, D.; Goldner, T.; Ruebsamen-Schaeff, H.; Zimmermann, H; In vitro and in vivo activities of the novel anticytomegalovirus compound AIC246. Antimicrob. Agents Chemother. 2010, 54, 1290–1297.
- Steffen Wildum; Holger Zimmermann; Peter Lischka; In Vitro Drug Combination Studies of Letermovir (AIC246, MK-8228) with Approved Anti-Human Cytomegalovirus (HCMV) and Anti-HIV Compounds in Inhibition of HCMV and HIV Replication. Antimicrobial Agents and Chemotherapy 2015, 59, 3140-3148, 10.1128/AAC.00114-15.
- Lauren Cherrier; Aasya Nasar; Kellie J. Goodlet; Michael D. Nailor; Sofya Tokman; Sunwen Chou; Emergence of letermovir resistance in a lung transplant recipient with ganciclovir-resistant cytomegalovirus infection. American Journal of Transplantation 2018, 18, 3060-3064, 10.1111/ajt.15135.
- Marja Van Zeijl; Jeanette Fairhurst; Ellen Z. Baum; Lei Sun; Thomas R. Jones; The Human Cytomegalovirus UL97 Protein Is Phosphorylated and a Component of Virions. Virology 1997, 231, 72-80, 10.1006/viro.1997.8523.
- Rike Webel; Morgan Hakki; Mark N. Prichard; Bill Rawlinson; Manfred Marschall; Sunwen Chou; Differential Properties of Cytomegalovirus pUL97 Kinase Isoforms Affect Viral Replication and Maribavir Susceptibility. Journal of Virology 2014, 88, 4776-4785, 10.1128/JVI.00192-14.
- Rike Webel; Jens Milbradt; Sabrina Auerochs; Vera Schregel; Christian Held; Katharina Nöbauer; Ebrahim Razzazi-Fazeli; Christophe Jardin; Thomas Wittenberg; Heinrich Sticht; Manfred Marschall; Two isoforms of the protein kinase pUL97 of human cytomegalovirus are differentially regulated in their nuclear translocation. Journal of General Virology 2010, 92, 638-649, 10.1099/vir.0.026799-0.
- Webel, R.; Solbak, S.M.; Held, C.; Milbradt, J.; Gross, A.; Eichler, J.; Wittenberg, T.; Jardin, C.; Sticht, H.; Fossen, T.; et al. Nuclear import of isoforms of the cytomegalovirus kinase pUL97 is mediated by differential activity of NLS1 and NLS2 both acting through classical importin-alpha binding. J. Gen. Virol. 2012, 93, 1756–1768.
- Mirjam Steingruber; Eileen Socher; Corina Hutterer; Rike Webel; Tim Bergbrede; Tihana Lenac; Heinrich Sticht; Manfred Marschall; The Interaction between Cyclin B1 and Cytomegalovirus Protein Kinase pUL97 is Determined by an Active Kinase Domain. Viruses 2015, 7, 4582-4601, 10.3390/v7082834.
- Adam Hume; R F Kalejta; Regulation of the retinoblastoma proteins by the human herpesviruses. Cell Division 2009, 4, 1, 10.1186/1747-1028-4-1.
- Edward Gershburg; Joseph S. Pagano; Conserved herpesvirus protein kinases. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2008, 1784, 203-212, 10.1016/j.bbapap.2007.08.009.
- D Michel; Thomas Mertens; The UL97 protein kinase of human cytomegalovirus and homologues in other herpesviruses: impact on virus and host. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2004, 1697, 169-180, 10.1016/j.bbapap.2003.11.022.
- Manfred Marschall; Matthias Stein-Gerlach; Martina Freitag; Regina Kupfer; Miriam Van Den Bogaard; Thomas Stamminger; Inhibitors of human cytomegalovirus replication drastically reduce the activity of the viral protein kinase pUL97. Journal of General Virology 2001, 82, 1439-1450, 10.1099/0022-1317-82-6-1439.
- Mark N. Prichard; Function of human cytomegalovirus UL97 kinase in viral infection and its inhibition by maribavir.. Reviews in Medical Virology 2009, 19, 215-29, 10.1002/rmv.615.
- Manfred Marschall; Sabine Feichtinger; Jens Milbradt; Regulatory Roles of Protein Kinases in Cytomegalovirus Replication. Advances in Applied Microbiology 2011, 80, 69-101, 10.1016/b978-0-12-385987-7.00004-x.
- Vera Schregel; Sabrina Auerochs; Ramona Jochmann; Katja Maurer; Thomas Stamminger; Manfred Marschall; Mapping of a self-interaction domain of the cytomegalovirus protein kinase pUL97. Journal of General Virology 2007, 88, 395-404, 10.1099/vir.0.82393-0.
- Mirjam Steingruber; Alexandra Kraut; Eileen Socher; Heinrich Sticht; Anna Reichel; Thomas Stamminger; Bushra Amin; Yohann Couté; Corina Hutterer; Manfred Marschall; Proteomic Interaction Patterns between Human Cyclins, the Cyclin-Dependent Kinase Ortholog pUL97 and Additional Cytomegalovirus Proteins. Viruses 2016, 8, 219, 10.3390/v8080219.
- Mirjam Steingruber; Lena Keller; Eileen Socher; Sabrina Ferre; Anne-Marie Hesse; Yohann Couté; Friedrich Hahn; Nicole Büscher; Bodo Plachter; Heinrich Sticht; Manfred Marschall; Manfrad Marschall; Cyclins B1, T1, and H differ in their molecular mode of interaction with cytomegalovirus protein kinase pUL97.. Journal of Biological Chemistry 2019, 294, 6188-6203, 10.1074/jbc.RA118.007049.
- Chad Kuny; Karen Chinchilla; Michael R. Culbertson; R F Kalejta; Cyclin-Dependent Kinase-Like Function Is Shared by the Beta- and Gamma- Subset of the Conserved Herpesvirus Protein Kinases. PLOS Pathogens 2010, 6, e1001092, 10.1371/journal.ppat.1001092.
- Laura Hertel; Sunwen Chou; Edward S. Mocarski; Viral and Cell Cycle–Regulated Kinases in Cytomegalovirus-Induced Pseudomitosis and Replication. PLOS Pathogens 2007, 3, e6, 10.1371/journal.ppat.0030006.
- Dana G. Wolf; Charmain Tan Courcelle; Mark N. Prichard; E S Mocarski; Distinct and separate roles for herpesvirus-conserved UL97 kinase in cytomegalovirus DNA synthesis and encapsidation. Proceedings of the National Academy of Sciences 2001, 98, 1895-1900, 10.1073/pnas.98.4.1895.
- Mark N. Prichard; William J. Britt; Shannon L. Daily; Caroll B. Hartline; Earl R. Kern; Human Cytomegalovirus UL97 Kinase Is Required for the Normal Intranuclear Distribution of pp65 and Virion Morphogenesis. Journal of Virology 2005, 79, 15494-15502, 10.1128/jvi.79.24.15494-15502.2005.
- Mark N. Prichard; Ning Gao; Sanju Jairath; Gilbert Mulamba; Paula Krosky; Donald M. Coen; Breck O. Parker; Gregory S. Pari; A Recombinant Human Cytomegalovirus with a Large Deletion in UL97 Has a Severe Replication Deficiency. Journal of Virology 1999, 73, 5663-5670, 10.1128/jvi.73.7.5663-5670.1999.
- Adam Hume; J. S. Finkel; Jeremy P. Kamil; D. M. Coen; M. R. Culbertson; R. F. Kalejta; Phosphorylation of Retinoblastoma Protein by Viral Protein with Cyclin-Dependent Kinase Function. Science 2008, 320, 797-799, 10.1126/science.1152095.
- Marco Thomas; Sabine Rechter; Jens Milbradt; Sabrina Auerochs; Regina Müller; Thomas Stamminger; Manfred Marschall; Cytomegaloviral protein kinase pUL97 interacts with the nuclear mRNA export factor pUL69 to modulate its intranuclear localization and activity. Journal of General Virology 2009, 90, 567-578, 10.1099/vir.0.005827-0.
- Sabine Feichtinger; Thomas Stamminger; Regina Müller; Laura Graf; Bert Klebl; Jan Eickhoff; Manfred Marschall; Recruitment of cyclin-dependent kinase 9 to nuclear compartments during cytomegalovirus late replication: importance of an interaction between viral pUL69 and cyclin T1. Journal of General Virology 2011, 92, 1519-1531, 10.1099/vir.0.030494-0.
- Laura Graf; Sabine Feichtinger; Zin Naing; Corina Hutterer; Jens Milbradt; Rike Webel; Sabrina Wagner; Gillian M. Scott; Stuart T. Hamilton; Bill Rawlinson; Thomas Stamminger; Marco Thomas; Manfred Marschall; New insight into the phosphorylation-regulated intranuclear localization of human cytomegalovirus pUL69 mediated by cyclin-dependent kinases (CDKs) and viral CDK orthologue pUL97. Journal of General Virology 2016, 97, 144-151, 10.1099/jgv.0.000337.
- Patrick König; Nicole Büscher; Mirjam Steingruber; Eileen Socher; Heinrich Sticht; Stefan Tenzer; Bodo Plachter; Manfred Marschall; Dynamic regulatory interaction between cytomegalovirus major tegument protein pp65 and protein kinase pUL97 in intracellular compartments, dense bodies and virions. Journal of General Virology 2017, 98, 2850-2863, 10.1099/jgv.0.000939.
- Sofia Hamirally; Jeremy P. Kamil; Yasmine M. Ndassa-Colday; Alison J. Lin; Wan Jin Jahng; Moon-Chang Baek; Sarah Noton; Laurie A. Silva; Martha Simpson-Holley; David M. Knipe; David E. Golan; Jarrod A. Marto; Nald M. Coen; Viral Mimicry of Cdc2/Cyclin-Dependent Kinase 1 Mediates Disruption of Nuclear Lamina during Human Cytomegalovirus Nuclear Egress. PLOS Pathogens 2009, 5, e1000275, 10.1371/journal.ppat.1000275.
- Manfred Marschall; Andrea Marzi; Patricia Aus Dem Siepen; Ramona Jochmann; Martina Kalmer; Sabrina Auerochs; Peter Lischka; Martina Leis; Thomas Stamminger; Cellular p32 Recruits Cytomegalovirus Kinase pUL97 to Redistribute the Nuclear Lamina. Journal of Biological Chemistry 2005, 280, 33357-33367, 10.1074/jbc.m502672200.
- Sabine Becke; Véronique Fabre-Mersseman; Steffi Aue; Sabrina Auerochs; Tina Sedmak; Uwe Wolfrum; Dennis Strand; Manfred Marschall; Bodo Plachter; Sabine Reyda; Modification of the major tegument protein pp65 of human cytomegalovirus inhibits virus growth and leads to the enhancement of a protein complex with pUL69 and pUL97 in infected cells. Journal of General Virology 2010, 91, 2531-2541, 10.1099/vir.0.022293-0.
- Valentina Dell'oste; Deborah Gatti; Francesca Gugliesi; Marco De Andrea; Mandar Bawadekar; Irene Lo Cigno; Matteo Biolatti; Marta Vallino; Manfred Marschall; Marisa Gariglio; Santo Landolfo; Innate Nuclear Sensor IFI16 Translocates into the Cytoplasm during the Early Stage of In Vitro Human Cytomegalovirus Infection and Is Entrapped in the Egressing Virions during the Late Stage. Journal of Virology 2014, 88, 6970-6982, 10.1128/JVI.00384-14.
- Jens Milbradt; Corina Hutterer; Hanife Bahsi; Sabrina Wagner; Eric Sonntag; Anselm H.C. Horn; Benedikt B. Kaufer; Yasuko Mori; Heinrich Sticht; Torgils Fossen; Manfred Marschall; The Prolyl Isomerase Pin1 Promotes the Herpesvirus-Induced Phosphorylation-Dependent Disassembly of the Nuclear Lamina Required for Nucleocytoplasmic Egress. PLOS Pathogens 2016, 12, e1005825, 10.1371/journal.ppat.1005825.
- Jens Milbradt; Alexandra Kraut; Corina Hutterer; Eric Sonntag; Cathrin Schmeiser; Myriam Ferro; Sabrina Wagner; Tihana Lenac; Claudia Claus; Sandra Pinkert; Stuart T. Hamilton; Bill Rawlinson; Heinrich Sticht; Yohann Couté; Manfred Marschall; Proteomic analysis of the multimeric nuclear egress complex of human cytomegalovirus.. Molecular & Cellular Proteomics 2014, 13, 2132-46, 10.1074/mcp.M113.035782.
- Sharma, M.; Bender, B.J.; Kamil, J.P.; Lye, M.F.; Pesola, J.M.; Reim, N.I.; Hogle, J.M.; Coen, D.M; Human cytomegalovirus UL97 phosphorylates the viral nuclear egress complex. J. Virol. 2015, 89, 523–534, 10.1128/jvi.02358-13.
- Moon-Chang Baek; Paula M Krosky; Angela Pearson; Donald M Coen; Phosphorylation of the RNA polymerase II carboxyl-terminal domain in human cytomegalovirus-infected cells and in vitro by the viral UL97 protein kinase. Virology 2004, 324, 184-193, 10.1016/j.virol.2004.03.015.
- Yasushi Kawaguchi; Tomio Matsumura; Bernard Roizman; K Hirai; Cellular Elongation Factor 1δ Is Modified in Cells Infected with Representative Alpha-, Beta-, or Gammaherpesviruses. Journal of Virology 1999, 73, 4456-4460.
- Manfred Marschall; Martina Freitag; Patricia Suchy; Daniel Romaker; Regina Kupfer; Miriam Hanke; Thomas Stamminger; The protein kinase pUL97 of human cytomegalovirus interacts with and phosphorylates the DNA polymerase processivity factor pUL44. Virology 2003, 311, 60-71, 10.1016/s0042-6822(03)00147-8.
- Walzer, S.A.; Egerer-Sieber, C.; Sticht, H.; Sevvana, M.; Hohl, K.; Milbradt, J.; Muller, Y.A.; Marschall, M; Faculty of 1000 evaluation for Crystal Structure of the Human Cytomegalovirus pUL50-pUL53 Core Nuclear Egress Complex Provides Insight into a Unique Assembly Scaffold for Virus-Host Protein Interactions.. J. Biol. Chem. 2015, 290, 27452–27458.
- Eric Sonntag; Jens Milbradt; Adriana Svrlanska; Hanife Strojan; Sigrun Häge; Alexandra Kraut; Anne-Marie Hesse; Bushra Amin; Uwe Sonnewald; Yohann Couté; Manfred Marschall; Protein kinases responsible for the phosphorylation of the nuclear egress core complex of human cytomegalovirus. Journal of General Virology 2017, 98, 2569-2581, 10.1099/jgv.0.000931.
- Eric Sonntag; Stuart T. Hamilton; Hanife Bahsi; Sabrina Wagner; Stipan Jonjić; Bill Rawlinson; Manfred Marschall; Jens Milbradt; Erratum to Cytomegalovirus pUL50 is the multi-interacting determinant of the core nuclear egress complex (NEC) that recruits cellular accessory NEC components. Journal of General Virology 2016, 97, 2461-2461, 10.1099/jgv.0.000599.
- Jens Milbradt; Rike Webel; Sabrina Auerochs; Heinrich Sticht; Manfred Marschall; Novel Mode of Phosphorylation-triggered Reorganization of the Nuclear Lamina during Nuclear Egress of Human Cytomegalovirus*. Journal of Biological Chemistry 2010, 285, 13979-13989, 10.1074/jbc.M109.063628.
- Yasushi Kawaguchi; Kentaro Kato; Michiko Tanaka; Mikiko Kanamori; Yukihiro Nishiyama; Yuji Yamanashi; Conserved protein kinases encoded by herpesviruses and cellular protein kinase cdc2 target the same phosphorylation site in eukaryotic elongation factor 1delta. J. Virol. 2003, 77, 2359–2368.
- Guillermo Ruiz-Carrascoso; Maria Pilar Romero-Gomez; Diego Plaza; Jesús Mingorance; Rapid detection and quantitation of ganciclovir resistance in cytomegalovirus quasispecies. Journal of Medical Virology 2013, 85, 1250-1257, 10.1002/jmv.23570.
- Sunwen Chou; Cytomegalovirus UL97 mutations in the era of ganciclovir and maribavir. Reviews in Medical Virology 2008, 18, 233-246, 10.1002/rmv.574.
- Manfred Marschall; Yves A. Muller; Benedikt Diewald; Heinrich Sticht; Jens Milbradt; The human cytomegalovirus nuclear egress complex unites multiple functions: Recruitment of effectors, nuclear envelope rearrangement, and docking to nuclear capsids. Reviews in Medical Virology 2017, 27, e1934, 10.1002/rmv.1934.
- Daniel Romaker; Vera Schregel; Katja Maurer; Sabrina Auerochs; Andrea Marzi; Heinrich Sticht; Manfred Marschall; Analysis of the Structure−Activity Relationship of Four Herpesviral UL97 Subfamily Protein Kinases Reveals Partial but not Full Functional Conservation†. Journal of Medicinal Chemistry 2006, 49, 7044-7053, 10.1021/jm060696s.
- Corina Hutterer; S. Hamilton; M. Steingruber; I. Zeitträger; H. Bahsi; N. Thuma; Z. Naing; Z. Örfi; László Őrfi; Eileen Socher; Heinrich Sticht; W. Rawlinson; S. Chou; V.J. Haupt; M. Marschall; Steingruber M; Zeitträger I; Chou S; Marschall M; The chemical class of quinazoline compounds provides a core structure for the design of anticytomegaloviral kinase inhibitors. Antiviral Research 2016, 134, 130-143, 10.1016/j.antiviral.2016.08.005.
- Moon-Chang Baek; Paula M Krosky; Nald M Coen; Relationship between autophosphorylation and phosphorylation of exogenous substrates by the human cytomegalovirus UL97 protein kinase. Journal of Virology 2002, 76, 11943–11952.
- Adam Oberstein; David H. Perlman; Thomas Shenk; Laura J. Terry; Human cytomegalovirus pUL97 kinase induces global changes in the infected cell phosphoproteome. PROTEOMICS 2015, 15, 2006-22, 10.1002/pmic.201400607.
- Michel, D.; Pavic, I.; Zimmermann, A.; Haupt, E.; Wunderlich, K.; Heuschmid, M.; Mertens, T; The UL97 gene product of human cytomegalovirus is an early-late protein with a nuclear localization but is not a nucleoside kinase. Journal of Virology 1996, 70, 6340–6346.
- Edward Littler; Amanda D. Stuart; Mark S. Chee; Human cytomegalovirus UL97 open reading frame encodes a protein that phosphorylates the antiviral nucleoside analogue ganciclovir. Nature 1992, 358, 160-162, 10.1038/358160a0.
- V. Sullivan; C. L. Talarico; S. C. Stanat; M. Davis; D. M. Coen; K. K. Biron; A protein kinase homologue controls phosphorylation of ganciclovir in human cytomegalovirus-infected cells. Nature 1992, 359, 85-85, 10.1038/359085a0.
- D Michel; I Pavić; A Zimmermann; E Haupt; K Wunderlich; M Heuschmid; T Mertens; The UL97 gene product of human cytomegalovirus is an early-late protein with a nuclear localization but is not a nucleoside kinase. Journal of Virology 1996, 70, 6340-6346.
- Christian Held; Rike Webel; Ralf Palmisano; Corina Hutterer; Manfred Marschall; Thomas Wittenberg; Using multi-channel level sets to measure the cytoplasmic localization of HCMV pUL97 in GFP-B-gal fusion constructs. Journal of Virological Methods 2014, 199, 61-67, 10.1016/j.jviromet.2013.12.009.
- Song Hee Lee; Katie Caviness; Emily R. Albright; Jeong-Hee Lee; Christopher B. Gelbmann; Mike Rak; Felicia Goodrum; R F Kalejta; Long and Short Isoforms of the Human Cytomegalovirus UL138 Protein Silence IE Transcription and Promote Latency. Journal of Virology 2016, 90, 9483-9494, 10.1128/JVI.01547-16.
- Sunwen Chou; Ronald J. Ercolani; Katayoun Derakhchan; Antiviral activity of maribavir in combination with other drugs active against human cytomegalovirus. Antiviral Research 2018, 157, 128-133, 10.1016/j.antiviral.2018.07.013.
- Katie Caviness; Farah Bughio; Lindsey B. Crawford; Daniel N. Streblow; Jay A. Nelson; Patrizia Caposio; Felicia Goodrum; Complex Interplay of the UL136 Isoforms Balances Cytomegalovirus Replication and Latency. mBio 2016, 7, 15, 10.1128/mBio.01986-15.
- Julia Sehl; Sandy Pörtner; Barbara G. Klupp; Harald Granzow; Kati Franzke; Jens P. Teifke; Thomas C. Mettenleiter; Roles of the Different Isoforms of the Pseudorabies Virus Protein Kinase pUS3 in Nuclear Egress. Journal of Virology 2020, -, -, 10.1128/jvi.02029-19.
- Charles Cunningham; Derek Gatherer; Birgitta Hilfrich; Katarina Baluchova; Derrick J. Dargan; Marian Thomson; Paul D. Griffiths; Gavin W. G. Wilkinson; Thomas F. Schulz; Andrew J. Davison; Sequences of complete human cytomegalovirus genomes from infected cell cultures and clinical specimens.. Journal of General Virology 2009, 91, 605-15, 10.1099/vir.0.015891-0.
- Nell S. Lurain; Adriana Weinberg; Clyde S. Crumpacker; Sunwen Chou; Sequencing of Cytomegalovirus UL97 Gene for Genotypic Antiviral Resistance Testing. Antimicrobial Agents and Chemotherapy 2001, 45, 2775-2780, 10.1128/aac.45.10.2775-2780.2001.
- Sabine Rechter; Gillian M. Scott; Jan Eickhoff; Katrin Zielke; Sabrina Auerochs; Regina Müller; Thomas Stamminger; Bill Rawlinson; Manfred Marschall; Cyclin-dependent Kinases Phosphorylate the Cytomegalovirus RNA Export Protein pUL69 and Modulate Its Nuclear Localization and Activity*S⃞. Journal of Biological Chemistry 2009, 284, 8605-8613, 10.1074/jbc.M805693200.
- Jeremy P. Kamil; Adam Hume; Igor Jurak; Karl Munger; Robert F. Kalejta; Nald M. Coen; Human papillomavirus 16 E7 inactivator of retinoblastoma family proteins complements human cytomegalovirus lacking UL97 protein kinase. Proceedings of the National Academy of Sciences 2009, 106, 16823-16828, 10.1073/pnas.0901521106.
- Corina Hutterer; Sebastian Karl Wandinger; Sabrina Wagner; Regina Müller; Thomas Stamminger; Isabel Zeitträger; Klaus Godl; Roland Baumgartner; Stefan Strobl; Manfred Marschall; Profiling of the kinome of cytomegalovirus-infected cells reveals the functional importance of host kinases Aurora A, ABL and AMPK. Antiviral Research 2013, 99, 139-148, 10.1016/j.antiviral.2013.04.017.
- Jens Milbradt; Sabrina Auerochs; Madhumati Sevvana; Yves A. Muller; Heinrich Sticht; Manfred Marschall; Specific Residues of a Conserved Domain in the N Terminus of the Human Cytomegalovirus pUL50 Protein Determine Its Intranuclear Interaction with pUL53*. Journal of Biological Chemistry 2012, 287, 24004-24016, 10.1074/jbc.M111.331207.
- Rachel B. Gill; Scott H. James; Mark N. Prichard; Human cytomegalovirus UL97 kinase alters the accumulation of CDK1. Journal of General Virology 2012, 93, 1743-1755, 10.1099/vir.0.039214-0.
- F M Jault; J M Jault; F Ruchti; E A Fortunato; C Clark; J Corbeil; D D Richman; D H Spector; Cytomegalovirus infection induces high levels of cyclins, phosphorylated Rb, and p53, leading to cell cycle arrest.. Journal of Virology 1995, 69, 6697-6704.
- Anokhi J. Kapasi; Deborah H. Spector; Inhibition of the Cyclin-Dependent Kinases at the Beginning of Human Cytomegalovirus Infection Specifically Alters the Levels and Localization of the RNA Polymerase II Carboxyl-Terminal Domain Kinases cdk9 and cdk7 at the Viral Transcriptosome. Journal of Virology 2008, 82, 394-407, 10.1128/jvi.01681-07.
- Veronica Sanchez; Anita K. McElroy; Deborah H. Spector; Mechanisms governing maintenance of Cdk1/cyclin B1 kinase activity in cells infected with human cytomegalovirus.. Journal of Virology 2003, 77, 13214–13224.
- Deborah H. Spector; Human cytomegalovirus riding the cell cycle. Medical Microbiology and Immunology 2015, 204, 409-419, 10.1007/s00430-015-0396-z.
- Sama Tamrakar; Anokhi J. Kapasi; Deborah H. Spector; Human Cytomegalovirus Infection Induces Specific Hyperphosphorylation of the Carboxyl-Terminal Domain of the Large Subunit of RNA Polymerase II That Is Associated with Changes in the Abundance, Activity, and Localization of cdk9 and cdk7. Journal of Virology 2005, 79, 15477-15493, 10.1128/jvi.79.24.15477-15493.2005.
- Mayuri Sharma; Jeremy P. Kamil; Margaret Coughlin; Natalia I. Reim; Nald M. Coen; Human Cytomegalovirus UL50 and UL53 Recruit Viral Protein Kinase UL97, Not Protein Kinase C, for Disruption of Nuclear Lamina and Nuclear Egress in Infected Cells. Journal of Virology 2013, 88, 249-262, 10.1128/jvi.02358-13.
- Kawaguchi, Y.; Matsumura, T.; Roizman, B.; Hirai, K. Cellular elongation factor 1delta is modified in cells infected with representative alpha-, beta-, or gammaherpesviruses. J. Virol. 1999, 73, 4456–4460.
- Tarin Bigley; Justin M. Reitsma; Shama P. Mirza; Scott S. Terhune; Human Cytomegalovirus pUL97 Regulates the Viral Major Immediate Early Promoter by Phosphorylation-Mediated Disruption of Histone Deacetylase 1 Binding. Journal of Virology 2013, 87, 7393-7408, 10.1128/JVI.02825-12.
- Satoko Iwahori; Angie C. Umaña; Halena R. VanDeusen; R F Kalejta; Human cytomegalovirus-encoded viral cyclin-dependent kinase (v-CDK) UL97 phosphorylates and inactivates the retinoblastoma protein-related p107 and p130 proteins. Journal of Biological Chemistry 2017, 292, 6583-6599, 10.1074/jbc.m116.773150.
- Paula M Krosky; Moon-Chang Baek; Wan Jin Jahng; Imma Barrera; Robert J Harvey; Karen K Biron; Nald M Coen; Phiroze Sethna; The human cytomegalovirus UL44 protein is a substrate for the UL97 protein kinase.. Journal of Virology 2003, 77, 7720–7727.
- Ramona Businger; Janina Deutschmann; Iris Gruska; Jens Milbradt; Lüder Wiebusch; Thomas Gramberg; Michael Schindler; Human cytomegalovirus overcomes SAMHD1 restriction in macrophages via pUL97. Nature Microbiology 2019, 4, 2260-2272, 10.1038/s41564-019-0557-8.
- Satoko Iwahori; R F Kalejta; Phosphorylation of transcriptional regulators in the retinoblastoma protein pathway by UL97, the viral cyclin-dependent kinase encoded by human cytomegalovirus. Virology 2017, 512, 95-103, 10.1016/j.virol.2017.09.009.
- Matteo Biolatti; Valentina Dell’Oste; Sara Pautasso; Jens Von Einem; Manfred Marschall; Bodo Plachter; Marisa Gariglio; Marco De Andrea; Santo Landolfo; Regulatory Interaction between the Cellular Restriction Factor IFI16 and Viral pp65 (pUL83) Modulates Viral Gene Expression and IFI16 Protein Stability. Journal of Virology 2016, 90, 8238-8250, 10.1128/JVI.00923-16.
- Santo Landolfo; Marco De Andrea; Valentina Dell’Oste; Francesca Gugliesi; Intrinsic host restriction factors of human cytomegalovirus replication and mechanisms of viral escape. World Journal of Virology 2016, 5, 87-96, 10.5501/wjv.v5.i3.87.
- Manfred Marschall; Matthias Stein-Gerlach; Martina Freitag; Regina Kupfer; Miriam Van Den Bogaard; Thomas Stamminger; Direct targeting of human cytomegalovirus protein kinase pUL97 by kinase inhibitors is a novel principle for antiviral therapy. Journal of General Virology 2002, 83, 1013-1023, 10.1099/0022-1317-83-5-1013.
- He, Z.; He, Y.S.; Kim, Y.; Chu, L.; Ohmstede, C.; Biron, K.K.; Coen, D.M; The human cytomegalovirus UL97 protein is a protein kinase that autophosphorylates on serines and threonines. J. Virol. 1997, 71, 405-411.
- Helmut Mett; Kerstin Hölscher; Heidrun Degen; Christina Esdar; Birgit Felden De Neumann; Birgit Flicke; Tatjana Freudenreich; Gaby Holzer; Sieglinde Schinzel; Thomas Stamminger; Matthias Stein-Gerlach; Manfred Marschall; Thomas Herget; Identification of Inhibitors for a Virally Encoded Protein Kinase by 2 Different Screening Systems: In Vitro Kinase Assay and In-Cell Activity Assay. Journal of Biomolecular Screening 2005, 10, 36-45, 10.1177/1087057104270269.
- Albert Zimmermann; Detlef Michel; Ivica Pavić; Walter Hampl; Anke Lüske; Johan Neyts; Erik De Clercq; Thomas Mertens; Phosphorylation of aciclovir, ganciclovir, penciclovir and S2242 by the cytomegalovirus UL97 protein: a quantitative analysis using recombinant vaccinia viruses. Antiviral Research 1997, 36, 35-42, 10.1016/s0166-3542(97)00034-x.
- Christine L. Talarico; Thimysta C. Burnette; Wayne H. Miller; Sheila L. Smith; Michelle G. Davis; Sylvia C. Stanat; Teresa I. Ng; Zuwen He; Donald M. Coen; Bernard Roizman; Karen K. Biron; Acyclovir Is Phosphorylated by the Human Cytomegalovirus UL97 Protein. Antimicrobial Agents and Chemotherapy 1999, 43, 1941-1946, 10.1128/aac.43.8.1941.
- Sunwen Chou; Laura C. Van Wechel; Gail I. Marousek; Cytomegalovirus UL97 Kinase Mutations That Confer Maribavir Resistance. The Journal of Infectious Diseases 2007, 196, 91-94, 10.1086/518514.
- G.M. Scott; M.A. Isaacs; F. Zeng; A.M. Kesson; Bill Rawlinson; Cytomegalovirus antiviral resistance associated with treatment induced UL97 (protein kinase) and UL54 (DNA polymerase) mutations. Journal of Medical Virology 2004, 74, 85-93, 10.1002/jmv.20150.
- D. G. Wolf; A. Honigman; J. Lazarovits; E. Tavor; Amos Panet; Characterization of the human cytomegalovirus UL97 gene product as a virion-associated protein kinase.. Archives of Virology 1998, 143, 1223-1232, 10.1007/s007050050370.
- Chevillotte, M.; Landwehr, S.; Linta, L.; Frascaroli, G.; Luske, A.; Buser, C.; Mertens, T.; von Einem, J; Major Tegument Protein pp65 of Human Cytomegalovirus Is Required for the Incorporation of pUL69 and pUL97 into the Virus Particle and for Viral Growth in Macrophages. Journal of Virology 2009, 83, 2480-2490, 10.1128/jvi.01818-08.
- Jens Milbradt; Eric Sonntag; Sabrina Wagner; Hanife Strojan; Christina Wangen; Tihana Lenac Rovis; Berislav Lisnic; Stipan Jonjić; Heinrich Sticht; William J. Britt; Ursula Schlötzer-Schrehardt; Manfred Marschall; Human Cytomegalovirus Nuclear Capsids Associate with the Core Nuclear Egress Complex and the Viral Protein Kinase pUL97. Viruses 2018, 10, 35, 10.3390/v10010035.
- Herget, T.; Marschall, M. Recent developments in anti-herpesviral combination therapy based on protein kinase inhibitors. In New Concepts of Antiviral Therapy; Springer: Boston, MA, USA, 2006; pp. 351–371.
- Albert Zimmermann; Heike Wilts; Martin Lenhardt; Meike Hahn; T Mertens; Indolocarbazoles exhibit strong antiviral activity against human cytomegalovirus and are potent inhibitors of the pUL97 protein kinase. Antiviral Research 2000, 48, 49-60, 10.1016/s0166-3542(00)00118-2.
- Slater, M.J.; Baxter, R.; Bonser, R.W.; Cockerill, S.; Gohil, K.; Parry, N.; Robinson, E.; Randall, R.; Yeates, C.; Snowden, W; et al. Synthesis of N-alkyl substituted indolocarbazoles as potent inhibitors of human cytomegalovirus replication. Bioorg. Med. Chem. Lett. 2001, 11, 1993-1995, 10.1016/s0960-894x(01)00352-3.
- Mark Schleiss; Jan Eickhoff; Sabrina Auerochs; Martina Leis; Silke Abele; Sabine Rechter; Yeon Choi; Jodi Anderson; Gillian Scott; Bill Rawlinson; Detlef Michel; Stephan Ensminger; Bert Klebl; Thomas Stamminger; Manfred Marschall; Protein kinase inhibitors of the quinazoline class exert anti-cytomegaloviral activity in vitro and in vivo. Antiviral Research 2008, 79, 49-61, 10.1016/j.antiviral.2008.01.154.
- Chou, S.; Marousek, G.I; Maribavir antagonizes the antiviral action of ganciclovir on human cytomegalovirus. Antimicrob. Agents Chemother. 2006, 50, 3470–3472.
- Graf, L.; Webel, R.; Wagner, S.; Hamilton, S.T.; Rawlinson, W.D.; Sticht, H.; Marschall, M; The Cyclin-Dependent Kinase Ortholog pUL97 of Human Cytomegalovirus Interacts with Cyclins. Viruses 2013, 5, 3213-3230, 10.3390/v5123213.
- Salvador Cazorla-Vázquez; Mirjam Steingruber; Manfred Marschall; Felix B. Engel; Human cytomegaloviral multifunctional protein kinase pUL97 impairs zebrafish embryonic development and increases mortality. Scientific Reports 2019, 9, 7219, 10.1038/s41598-019-43649-x.
- Kendra Leigh; Mayuri Sharma; My Sam Mansueto; Andras Boeszoermenyi; David Filman; James Hogle; Gerhard Wagner; Nald M. Coen; Haribabu Arthanari; Structure of a herpesvirus nuclear egress complex subunit reveals an interaction groove that is essential for viral replication. Proceedings of the National Academy of Sciences 2015, 112, 9010-9015, 10.1073/pnas.1511140112.
- Ming F Lye; Mayuri Sharma; Kamel El Omari; David Filman; Jonathan P Schuermann; James Hogle; Donald M Coen; Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. The EMBO Journal 2015, 34, 2937-2952, 10.15252/embj.201592651.
- Yves A. Muller; Sigrun Häge; Sewar Alkhashrom; Tobias Höllriegl; Sebastian Weigert; Simon Dolles; Kerstin Hof; Sascha A. Walzer; Claudia Egerer-Sieber; Marcus Conrad; Stephanie Holst; Josephine Lösing; Eric Sonntag; Heinrich Sticht; Jutta Eichler; Manfred Marschall; High-resolution crystal structures of two prototypical β- and γ-herpesviral nuclear egress complexes unravel the determinants of subfamily specificity. Journal of Biological Chemistry 2020, 295, 3189-3201, 10.1074/jbc.ra119.011546.
- P. Dal Monte; S. Pignatelli; Nicoletta Zini; N. M. Maraldi; E. Perret; M. C. Prévost; M. P. Landini; Analysis of intracellular and intraviral localization of the human cytomegalovirus UL53 protein. Journal of General Virology 2002, 83, 1005-1012, 10.1099/0022-1317-83-5-1005.
- Peter Lischka; Olaf Rosorius; Erik Trommer; Thomas Stamminger; A novel transferable nuclear export signal mediates CRM1-independent nucleocytoplasmic shuttling of the human cytomegalovirus transactivator protein pUL69. The EMBO Journal 2001, 20, 7271-7283, 10.1093/emboj/20.24.7271.
- Thomas, M.; Müller, R.; Horn, G.; Bogdanow, B.; Imami, K.; Milbradt, J.; Steingruber, M.; Marschall, M.; Schilling, E.M.; Fossen, T; et al. Phosphosite analysis of the cytomegaloviral mRNA export factor pUL69 reveals serines with critical importance for recruitment of cellular proteins Pin1 and UAP56/URH49. J. Virol. 2020, 94, e02151-19.
- Mayuri Sharma; Jeremy P. Kamil; Donald M. Coen; Preparation of the Human Cytomegalovirus Nuclear Egress Complex and Associated Proteins.. Part B: Numerical Computer Methods 2015, 569, 517-26, 10.1016/bs.mie.2015.08.020.
- Walter Muranyi; Jürgen Haas; Markus Wagner; Georg Krohne; Ulrich H. Koszinowski; Cytomegalovirus Recruitment of Cellular Kinases to Dissolve the Nuclear Lamina. Science 2002, 297, 854-857, 10.1126/science.1071506.
- Janina Deutschmann; Andrea Schneider; Iris Gruska; Barbara Vetter; Minique Thomas; Melissa Kießling; Sabine Wittmann; Alexandra Herrmann; Michael Schindler; Jens Milbradt; Nerea Ferreirós; Thomas H. Winkler; Lüder Wiebusch; Thomas Gramberg; A viral kinase counteracts in vivo restriction of murine cytomegalovirus by SAMHD1. Nature Microbiology 2019, 4, 2273-2284, 10.1038/s41564-019-0529-z.
- Sunwen Chou; Laura C. Van Wechel; Gail I. Marousek; Effect of Cell Culture Conditions on the Anticytomegalovirus Activity of Maribavir. Antimicrobial Agents and Chemotherapy 2006, 50, 2557-2559, 10.1128/aac.00207-06.
- Veronica Sanchez; Deborah H. Spector; Cyclin-Dependent Kinase Activity Is Required for Efficient Expression and Posttranslational Modification of Human Cytomegalovirus Proteins and for Production of Extracellular Particles. Journal of Virology 2006, 80, 5886-5896, 10.1128/JVI.02656-05.
- Peter D Adams; Regulation of the retinoblastoma tumor suppressor protein by cyclin/cdks. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 2001, 1471, M123-M133, 10.1016/s0304-419x(01)00019-1.
- Mark N. Prichard; Elizabeth Sztul; Shannon L. Daily; Amie Perry; Samuel L. Frederick; Rachel B. Gill; Caroll B. Hartline; Daniel N. Streblow; Susan M. Varnum; Richard D. Smith; Earl R. Kern; Human Cytomegalovirus UL97 Kinase Activity Is Required for the Hyperphosphorylation of Retinoblastoma Protein and Inhibits the Formation of Nuclear Aggresomes▿. Journal of Virology 2008, 82, 5054-5067, 10.1128/JVI.02174-07.
- Malumbres, M; Cyclin-dependent kinases. Genome Biol. 2014, 15, 122.
- Whittaker, S.R.; Mallinger, A.; Workman, P.; Clarke, P.A; Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol. Ther. 2017, 173, 83–105.
- Hung-Chuan Chiu; Wei-Ru Huang; Tsai-Ling Liao; Pei-I Chi; Brent L. Nielsen; Jyung-Hurng Liu; Hung-Jen Liu; Mechanistic insights into avian reovirus p17-modulated suppression of cell cycle CDK-cyclin complexes and enhancement of p53 and cyclin H interaction.. Journal of Biological Chemistry 2018, 293, 12542-12562, 10.1074/jbc.RA118.002341.
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Manning, G.; Morgan, D.O.; Tsai, L.H.; Wolgemuth, D.J; Cyclin-dependent kinases: A family portrait. Nat. Cell Biol. 2009, 11, 1275–1276.
- Jean Gautier; Jeremy Minshull; Manfred Lohka; Michael Glotzer; Tim Hunt; James L. Maller; Cyclin is a component of maturation-promoting factor from Xenopus. Cell 1990, 60, 487-494, 10.1016/0092-8674(90)90599-a.
- Lolli, G.; Lowe, E.D.; Brown, N.R.; Johnson, L.N; The crystal structure of human CDK7 and its protein recognition properties. Structure 2004, 12, 2067–2079.
- Fumiko Toyoshima-Morimoto; Eri Taniguchi; Nobuko Shinya; Akihiro Iwamatsu; Eisuke Nishida; Polo-like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature 2001, 410, 215-220, 10.1038/35065617.
- Yuan, J.; Eckerdt, F.; Bereiter-Hahn, J.; Kurunci-Csacsko, E.; Kaufmann, M.; Strebhardt, K; Cooperative phosphorylation including the activity of polo-like kinase 1 regulates the subcellular localization of cyclin B1. Oncogene 2002, 21, 8282–8292.
- Tarin Bigley; Justin M. Reitsma; Scott S. Terhune; Antagonistic Relationship between Human Cytomegalovirus pUL27 and pUL97 Activities during Infection. Journal of Virology 2015, 89, 10230-10246, 10.1128/JVI.00986-15.
- Sarah Garrett; William A. Barton; Ronald Knights; Pei Jin; David O. Morgan; Robert P. Fisher; Reciprocal Activation by Cyclin-Dependent Kinases 2 and 7 Is Directed by Substrate Specificity Determinants outside the T Loop. Molecular and Cellular Biology 2001, 21, 88-99, 10.1128/mcb.21.1.88-99.2001.
- Jae Bum Kim; Phillip A. Sharp; Positive Transcription Elongation Factor b Phosphorylates hSPT5 and RNA Polymerase II Carboxyl-terminal Domain Independently of Cyclin-dependent Kinase-activating Kinase. Journal of Biological Chemistry 2001, 276, 12317-12323, 10.1074/jbc.m010908200.
- A-M. Martinez; Mohammad Afshar; Francois Martin; Jean‐Claude Cavadore; Jean‐Claude Labbé; Marcel Dorée; Dual phosphorylation of the T-loop in cdk7: its role in controlling cyclin H binding and CAK activity.. The EMBO Journal 1997, 16, 343-354, 10.1093/emboj/16.2.343.
- Mbonye, U.; Wang, B.; Gokulrangan, G.; Shi, W.; Yang, S.; Karn, J. Cyclin-dependent kinase 7 (CDK7)-mediated phosphorylation of the CDK9 activation loop promotes P-TEFb assembly with Tat and proviral HIV reactivation. J. Biol. Chem. 2018, 293, 10009–10025.
- Rajesh Ramakrishnan; Andrew P. Rice; Cdk9 T-loop phosphorylation is regulated by the calcium signaling pathway. Journal of Cellular Physiology 2012, 227, 609-617, 10.1002/jcp.22760.
- Alicia A. Russo; Philip D. Jeffrey; Nikola P. Pavletich; Structural basis of cyclin-dependent kinase activation by phosphorylation. Nature Structural & Molecular Biology 1996, 3, 696-700, 10.1038/nsb0896-696.
- Oleg Timofeev; Onur Cizmecioglu; Florian Settele; Tore Kempf; Ingrid Hoffmann; Cdc25 Phosphatases Are Required for Timely Assembly of CDK1-Cyclin B at the G2/M Transition*. Journal of Biological Chemistry 2010, 285, 16978-16990, 10.1074/jbc.M109.096552.
- J A Lees; K J Buchkovich; D R Marshak; C W Anderson; E Harlow; The retinoblastoma protein is phosphorylated on multiple sites by human cdc2.. The EMBO Journal 1991, 10, 4279–4290.
- Cristiano Simone; Luigi Bagella; Cristiana Bellan; Antonio Giordano; Physical interaction between pRb and cdk9/cyclinT2 complex. Oncogene 2002, 21, 4158-4165, 10.1038/sj.onc.1205511.
- L J Ko; S Y Shieh; X Chen; L Jayaraman; K Tamai; Y Taya; C Prives; Z Q Pan; p53 is phosphorylated by CDK7-cyclin H in a p36MAT1-dependent manner.. Molecular and Cellular Biology 1997, 17, 7220-7229, 10.1128/mcb.17.12.7220.
- Danupon Nantajit; Ming Fan; Nadire Duru; Yunfei Wen; John C. Reed; Jian Jian Li; Cyclin B1/Cdk1 Phosphorylation of Mitochondrial p53 Induces Anti-Apoptotic Response. PLOS ONE 2010, 5, e12341, 10.1371/journal.pone.0012341.
- Senthil K. Radhakrishnan; Andrei L. Gartel; CDK9 Phosphorylates p53 on Serine Residues 33, 315 and 392. Cell Cycle 2006, 5, 519-521, 10.4161/cc.5.5.2514.
- Rebecca Heald; Frank McKeon; Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell 1990, 61, 579-589, 10.1016/0092-8674(90)90470-y.
- Gary Ward; Marc W. Kirschner; Identification of cell cycle-regulated phosphorylation sites on nuclear lamin C. Cell 1990, 61, 561-577, 10.1016/0092-8674(90)90469-u.
- Kira Glover-Cutter; Stephane LaRochelle; Benjamin Erickson; Chao Zhang; Kevan Shokat; Robert P. Fisher; David Bentley; TFIIH-Associated Cdk7 Kinase Functions in Phosphorylation of C-Terminal Domain Ser7 Residues, Promoter-Proximal Pausing, and Termination by RNA Polymerase II. Molecular and Cellular Biology 2009, 29, 5455-5464, 10.1128/mcb.00637-09.
- Graziano Lolli; Binding to DNA of the RNA-polymerase II C-terminal domain allows discrimination between Cdk7 and Cdk9 phosphorylation.. Nucleic Acids Research 2009, 37, 1260-8, 10.1093/nar/gkn1061.
- Alexandra Cribier; Benjamin Descours; Ana Luiza Chaves Valadão; Nadine Laguette; Monsef Benkirane; Phosphorylation of SAMHD1 by Cyclin A2/CDK1 Regulates Its Restriction Activity toward HIV-1. Cell Reports 2013, 3, 1036-1043, 10.1016/j.celrep.2013.03.017.
- Tommy E. White; Alberto Brandariz-Nuñez; Jose Carlos Valle-Casuso; Sarah Amie; Laura Anh Nguyen; Baek Kim; Marina Tuzova; Felipe Diaz-Griffero; The retroviral restriction ability of SAMHD1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation.. Cell Host & Microbe 2013, 13, 441–451, 10.1016/j.chom.2013.03.005.
- Zhang, K.; Lv, D.-W.; Li, R; Conserved Herpesvirus Protein Kinases Target SAMHD1 to Facilitate Virus Replication. Cell Rep. 2019, 28, 449–459.
- Brian G. Gentry; Sara N. Gentry; Trachette L. Jackson; Jiri Zemlicka; John C. Drach; Phosphorylation of antiviral and endogenous nucleotides to di- and triphosphates by guanosine monophosphate kinase. Biochemical Pharmacology 2011, 81, 43-49, 10.1016/j.bcp.2010.09.005.
- Biron, K.K.; Harvey, R.J.; Chamberlain, S.C.; Good, S.S.; Smith, A.A., 3rd; Davis, M.G.; Talarico, C.L.; Miller, W.H.; Ferris, R.; Dornsife, R.E; et al. Potent and selective inhibition of human cytomegalovirus replication by 1263W94, a benzimidazole L-riboside with a unique mode of action.. Antimicrob. Agents Chemother. 2002, 46, 2365–2372.
- Marschall, M.; Strojan, H.; Kiener, R.; Wangen, C.; Sonntag, E.; Muller, R.; Zeittrager, I.; Wagner, S.; Stamminger, T.; Milbradt, J.; et al. Differential upregulation of host cell protein kinases by the replication of alpha-, beta- and gamma-herpesviruses provides a signature of virus-specific signalling. J. Gen. Virol. 2020, 101, 284–289.
- Beatrice Mercorelli; Elisa Sinigalia; Arianna Loregian; Giorgio Palù; Human cytomegalovirus DNA replication: antiviral targets and drugs. Reviews in Medical Virology 2008, 18, 177-210, 10.1002/rmv.558.
- Thomas Herget; Martina Freitag; Monika Morbitzer; Regina Kupfer; Thomas Stamminger; Manfred Marschall; Novel Chemical Class of pUL97 Protein Kinase-Specific Inhibitors with Strong Anticytomegaloviral Activity. Antimicrobial Agents and Chemotherapy 2004, 48, 4154-4162, 10.1128/aac.48.11.4154-4162.2004.
- Sunwen Chou; Ronald J. Ercolani; Gail Marousek; Terry L. Bowlin; Cytomegalovirus UL97 Kinase Catalytic Domain Mutations That Confer Multidrug Resistance. Antimicrobial Agents and Chemotherapy 2013, 57, 3375-3379, 10.1128/AAC.00511-13.
- Biron, K.K. Maribavir: A Promising New Antiherpes Therapeutic Agent. In New Concepts of Antiviral Therapy; Springer: Boston, MA, USA, 2006.
- George W Koszalka; Nelson W Johnson; Steven S Good; Leslie Boyd; Stanley C Chamberlain; LeRoy B Townsend; John C. Drach; Karen K Biron; Preclinical and toxicology studies of 1263W94, a potent and selective inhibitor of human cytomegalovirus replication. Antimicrobial Agents and Chemotherapy 2002, 46, 2373–2380.
- Jacob P. Lalezari; Judith A. Aberg; Laurene H. Wang; Mary Beth Wire; Richard Miner; Wendy Snowden; Christine L. Talarico; Shuching Shaw; Mark A. Jacobson; W. Lawrence Drew; Phase I Dose Escalation Trial Evaluating the Pharmacokinetics, Anti-Human Cytomegalovirus (HCMV) Activity, and Safety of 1263W94 in Human Immunodeficiency Virus-Infected Men with Asymptomatic HCMV Shedding. Antimicrobial Agents and Chemotherapy 2002, 46, 2969-2976, 10.1128/aac.46.9.2969-2976.2002.
- Joseph D. Ma; Anne N. Nafziger; Stephen A. Villano; Andrea Gaedigk; Joseph S. Bertino; Maribavir Pharmacokinetics and the Effects of Multiple-Dose Maribavir on Cytochrome P450 (CYP) 1A2, CYP 2C9, CYP 2C19, CYP 2D6, CYP 3A, N-Acetyltransferase-2, and Xanthine Oxidase Activities in Healthy Adults. Antimicrobial Agents and Chemotherapy 2006, 50, 1130-1135, 10.1128/aac.50.4.1130-1135.2006.
- Marty, F.M.; Boeckh, M; Maribavir and human cytomegalovirus—what happened in the clinical trials and why might the drug have failed?. Curr. Opin. Virol. 2011, 1, 555-562, 10.1016/j.coviro.2011.10.011.
- David L Evers; Gloria Komazin; Dongjin Shin; Debbie D Hwang; LeRoy B Townsend; John C. Drach; Interactions among antiviral drugs acting late in the replication cycle of human cytomegalovirus. Antiviral Research 2002, 56, 61-72, 10.1016/s0166-3542(02)00094-3.
- James, S.H.; Hartline, C.B.; Harden, E.A.; Driebe, E.M.; Schupp, J.M.; Engelthaler, D.M.; Keim, P.S.; Bowlin, T.L.; Kern, E.R.; Prichard, M.N; et al. Cyclopropavir inhibits the normal function of the human cytomegalovirus UL97 kinase. Antimicrob. Agents Chemother. 2011, 55, 4682–4691.
- Sunwen Chou; Rachel H. Waldemer; Anne E. Senters; Kevin S. Michels; George W. Kemble; Richard C. Miner; W. Lawrence Drew; Cytomegalovirus UL97 Phosphotransferase Mutations That Affect Susceptibility to Ganciclovir. The Journal of Infectious Diseases 2002, 185, 162-169, 10.1086/338362.
- McSharry, J.J.; McDonough, A.; Olson, B.; Talarico, C.; Davis, M.; Biron, K.K. Inhibition of ganciclovir-susceptible and -resistant human cytomegalovirus clinical isolates by the benzimidazole L-riboside 1263W94. Clin. Diagn. Lab. Immunol. 2001, 8, 1279–1281.
- Stephanie L. Williams; Caroll B. Hartline; Nicole L. Kushner; Emma A. Harden; Deborah J. Bidanset; John C. Drach; Leroy B. Townsend; Mark R. Underwood; Karen K. Biron; Earl R. Kern; In Vitro Activities of Benzimidazole d- and l-Ribonucleosides against Herpesviruses. Antimicrobial Agents and Chemotherapy 2003, 47, 2186-2192, 10.1128/aac.47.7.2186-2192.2003.
- Sunwen Chou; Gail I. Marousek; Anne E. Senters; Michelle G. Davis; Karen K. Biron; Mutations in the Human Cytomegalovirus UL27 Gene That Confer Resistance to Maribavir. Journal of Virology 2004, 78, 7124-7130, 10.1128/jvi.78.13.7124-7130.2004.
- Gloria Komazin; Leroy B. Townsend; John C. Drach; Role of a Mutation in Human Cytomegalovirus Gene UL104 in Resistance to Benzimidazole Ribonucleosides. Journal of Virology 2004, 78, 710-715, 10.1128/jvi.78.2.710-715.2004.
- Mark N. Prichard; Debra C. Quenelle; Deborah J Bidanset; Gloria Komazin; Sunwen Chou; John C. Drach; Earl R. Kern; Human cytomegalovirus UL27 is not required for viral replication in human tissue implanted in SCID mice. Virology Journal 2006, 3, 18, 10.1186/1743-422X-3-18.