Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Mario I Vega and Version 2 by Rita Xu.
Resistance to therapy and disease progression are the main causes of mortality in most cancers. In particular, the development of resistance is an important limitation affecting the efficacy of therapeutic alternatives for cancer, including chemotherapy, radiotherapy, and immunotherapy. Signaling pathways are largely responsible for the mechanisms of resistance to cancer treatment and progression, and multiple myeloma is no exception. p38 mitogen-activated protein kinase (p38) is downstream of several signaling pathways specific to treatment resistance and progression.
- multiple myeloma
- p38
- transcriptional factor
- cancer progression
1. Introduction
Multiple myeloma (MM) is a hematologic malignancy that remains incurable, as most patients eventually relapse or become refractory to treatment [1]. Even though recent treatments have been improved, resistance to treatment persists in MM [2]. New therapeutic agents have recently been developed for the treatment of resistant or refractory MM, including immunomodulatory agents, proteasome inhibitors, monoclonal antibodies, and therapies directed at molecular targets in different signaling pathways. These therapeutic alternatives have shown relative antitumor activity in resistant and refractory MM, and recent combinations of these therapies have shown clinical effectiveness [3]. Recent studies have focused on therapies directed at molecular targets in refractory or relapsed MM, which are based on signaling pathways specifically activated in tumor cells, as is the case for p38 [3]. Mitogen-activated protein kinases (MAPK) allow cells to respond to a wide variety of stimuli and signals such as DNA damage or inflammatory cytokine activity, and extracellular stimuli such as oxidative and osmotic stress and heat shock [4][5][4,5]. p38 has four well-characterized isoforms: p38α, β, γ, and δ [6]. These four isoforms participate in the translation of signals that play a very important role in different cellular processes, such as cell proliferation, differentiation, glucose metabolism, and lipid secretion; senescence; the stress response; apoptosis; autophagy; and cell migration [7][8][9][7,8,9]. Depending on the context, p38 can act as a tumor promoter or tumor suppressor [7][10][7,10]. p38 is constitutively activated in MM and has been implicated in osteoclast and osteoblast activity and bone destruction [11]. Recent studies have shown that treatment with bortezomib can induce p38 activation, and this activation may be related to chemoresistance [12][13][12,13]. However, other studies show that p38 activation is associated with the induction of apoptosis and autophagy in MM [14].
2. p38 as a Molecular Target in Multiple Myeloma Therapy
Multiple studies have shown that interleukin-6 (IL-6) is an important growth factor involved in the physiopathogenesis of multiple myeloma [15][98], and through an autocrine/paracrine form, promotes the survival and proliferation of myeloma cells. It has also been shown that growth factors such as granulocyte colony-stimulating factor (G-CSF), as well as IL-10, may also play a role in MM cell lines [16][17][99,100]. IL-6 can induce the activation of different signaling pathways that may be involved in the physiopathogenesis of this disease. As already mentioned, these include the Jak-Stat, Ras/Raf/Mek/Erk, and AKT pathways [16][99], which are essential for MM cell proliferation. Additionally, it has been shown that mutations in Ras (N-Ras and K-Ras) lead to constitutive activation that also influences a more malignant phenotype in patients with MM [18][101]. This Ras activation-dependent proliferation may be independent of the Mek/Erk pathway, which makes the use of inhibitors of this pathway unfeasible as a therapeutic alternative in cases of MM with malignant phenotypes [19][102]. Studies have shown that heme oxygenase-1 (HO-1), an enzyme that provides potent cytoprotection, cell proliferation, and drug resistance [20][103], is expressed and correlates with the expression of IL-6 in the bone marrow microenvironment of MM patients and autocrine IL-6 in MM patient cells, and both HO-1 and IL-6 have been associated with disease severity in MM [21][104]. The increased expression of HO-1 can induce the increased expression of IL-6 through p38 and other MAPKs. Therefore, the chemical inhibition of p38 significantly inhibits the expression of IL-6 mediated by HO-1, and this may have therapeutic importance in MM. Other growth factors, such as insulin-like growth factor 1 (IGF-1), have also been shown to induce activation of the Akt and Mek/Erk pathways in MM cell lines, where it appears that the PI-3/Akt pathway plays a major role [16][17][99,100]. As already mentioned in the section on chemical inhibitors for p38, certain studies have associated the activity of the p38 pathway with the pathophysiology of MM, since the inhibition of this pathway by the chemical inhibitor VX-745 induces inhibited IL-6 secretion in MM cells and inhibited cell proliferation. Thus, the use of a combination of bortezomib with SCIO-469 or BIRB-796, as well LY2228820, also inhibits osteoclastogenesis [12][22][23][12,89,94]. Therefore, it is believed that p38 indirectly participates in the physiopathogenesis of MM in an indirect way, and this inhibition may have therapeutic implications in the treatment of MM [12]. Studies have shown that treatment with bortezomib induces the induction of apoptosis through p38 inhibition, which is mediated by the increased expression of p53 and decreased expression of Bcl-XL and Mcl-1 [24][85]. It has also been reported that p38 inhibition decreases the expression of IL-6 and VGEF by BM stromal cells, resulting in the inhibition of MM cell proliferation and adhesion [25][87], thus decreasing the tumor burden and angiogenesis in murine models of MM [26][27][84,86]. The interaction between bone marrow stromal cells (BMSCs) and multiple myeloma cells is very important in the pathogenesis of MM through the secretion of growth factors, cytokines, and extracellular vesicles. Exosomes appear to play an important role in the communication between BMSCs and MM cells through the transfer of cytokines between other molecules. In an in vivo model, it was shown that BMSC and MM cells could exchange exosomes carrying certain cytokines, inducing increased growth of MM cells and resistance to bortezomib [28][105]. Thus, they also induce the activation of several important pathways for survival, including p38. Therefore, these studies suggest that p38 inhibitors may have an important therapeutic role in preventing the effect of exosome-mediated BMSCs on the proliferation, migration, survival, and drug resistance of MM cells. Recent studies have shown that the TAK1 inhibitor induces the inhibition of proliferation and apoptosis in MM cells through the constitutive or melphalan-mediated inhibition of TAK1, NF-kB, and p38 [29][106]. Additional studies have shown that the selective TAK1 inhibitor 5Z-7-oxozeaene shows synergistic potential with bortezomib, inducing increased inhibition of proliferation and apoptosis [30][107]. This biological activity is related to the inhibition of TAK1, which induces the inhibition of JNK, MAPK p38, and Erk, which are activated by TAK1 in MM cell lines. Therefore, various studies have reported the use of chemical inhibitors of growth signaling pathways in MM as a therapeutic alternative, such as inhibitors of the Stat3- and Erk2-dependent IL-6 activation pathways. Another candidate is the proteasome inhibitor PS-341, which has been shown to activate the JNK pathway and inhibit the Erk1/Erk2-dependent pathway [31][79]. The possibility that MAPK pathways participate in the growth and physiopathogenesis of MM in general has led to the development of studies with the aim of using MAPK inhibitors as potential therapeutic agents alone or in combination with chemotherapy or other conventional or novel therapies. MAPKAPK2 (MK2) is the direct substrate of MAPK p38. Recent studies have shown that MK2 plays an important role in the pathophysiology of MM [32][108]. These studies suggest that using p38 inhibitors that affect MK2 or direct MK2 inhibitors could be an important therapeutic alternative for MM. One study reported that rafoxanide, an antiparasitic, is capable of inducing mitochondria-dependent proliferation inhibition and apoptosis in MM cells [33][109]. This is a consequence of the inhibition of p38 and Stat-1. Therefore, non-specific inhibitors that impact the p38 pathway may also be therapeutic alternatives for MM. It has recently been reported that p38 also regulates the expression of the NKG2D and DNAM-1 ligands in MM cells in a drug-dependent manner, sensitizing them to the induction of death by NK cells [34][110]. This has been determined to be mediated by the p38 activation of transcription factor E2F1. These results suggest that p38 inhibition could be a therapeutic alternative in cases of immunoresistance in MM mediated by NK cells. Spleen tyrosine kinase (Syk) is an intracellular enzyme that plays an important role in the activation of B cells or T cell receptors. Studies have shown that Syk inhibitors can inhibit proliferation and induce apoptosis in these kinase cells in MM, where inhibition of the p38 pathway has been shown to be one of the consequences of such treatment [35][71]. The combination of the Syk inhibitor and p38 inhibitor results in the increased induction of apoptosis in MM cell lines. Trifluoperazine is a drug used in psychosis, but recent studies have shown that it inhibits tumor growth in different types of cancer, including MM cells [36][111]. Additional studies found that this drug in combination with bortezomib had a cytotoxic effect in in vitro and in vivo models of MM by inducing proliferation inhibition and apoptosis mediated by p38 inhibition [36][111]. In vivo and in vitro studies have shown that celastrol, a pentacyclic nortriterpen quinone, has an antitumor effect in MM and other types of cancer [37][112]. Recent studies have shown that celastrol induces apoptosis in MM cell lines alone or in combination with bortezomib, and that the cytotoxic effect is also associated with inhibition of the IRAK4/ERK/p38 pathway [38][113]. Tris(dibenzylideneacetone)dipalladium (Tris DBA) is a small molecule of the palladium complex that induces inhibited cell growth and proliferation in multiple myeloma cells, either alone or in combination with bortezomib. This effect is a consequence of the inhibition of downstream p38 signaling [39][114]. Interestingly, Tris DBA reverses hypoxia-mediated drug resistance by inhibiting p38 and Hif-1α [40][115]. The cell-derived protein kinase T-LAK/PDZ-binding kinase (TOPK/PBK) has been proposed as a potential therapeutic target due to its low expression in most normal tissues and high expression in various tumors, including MM. Recent studies have reported that OTS514, a TOPK/PBK inhibitor, had a cytotoxic effect on MM cell lines [41][116]. OTS514 induces apoptosis by inhibiting FOXM1, Akt, and p38. Histone deacetylases are potential therapeutic targets in hematological malignancies. Recent studies have shown that a new histone deacetylase inhibitor induces cytotoxicity in MM cells via apoptosis and was able to induce cytotoxicity in myeloma cells co-cultured with bone mesenchymal stromal cells and osteoclasts previously treated with the inhibitor [42][117]. This inhibitor was found to suppress osteoclast differentiation and resorption in vitro by inhibiting ERK, p38, AKT, and JNK, which prevented MM-associated bone loss in an in vivo model. These results support the idea that inhibitors of MAPKs, and in particular, p38, may have potential clinical use in multiple myeloma treatment in the near future. Various groups have shown that p38 is constitutively active in myeloma cells and that this leads to osteolytic bone destruction [11][25][31][11,79,87]. Therefore, treatment with a p38 inhibitor decreased tumor burden and bone lesions in a murine myeloma model, prolonging survival [27][86]. Additionally, the inhibition of p38 has an important effect on reducing osteolytic bone lesions induced by MM, reducing osteoclastogenesis and improving osteoblastogenesis [11]. The important role of p38 in bone damage induced by MM was confirmed using shRNA specific to p38α in vitro, where lower bone resorption associated with the knockdown effect of p38 was observed. Therefore, p38 inhibition in special p38α/β inhibitors is positioned as an important therapeutic alternative in the prevention of osteolytic bone lesions caused by MM [11][43][11,118]. The CXCR4 chemokine receptor is expressed in a wide variety of hematological malignancies, including MM [44][119], and in conjunction with its ligand SDF-1, it plays an important role in cancer progression. Studies in which researchers generated a humanized mAb, hz515H7, which binds to human CXCR4, showed that this binding inhibits the signaling pathway induced by SDF-1, reducing the phosphorylation of Akt, Erk1/2, and p38, which strongly inhibits cell migration and proliferation [45][120]. Therefore, the inhibition of the p38 signaling pathway as part of the mechanism of action of humanized mAbs used therapeutically in MM may be important in their therapeutic efficacy. Additionally, studies using gambogic acid (GA), a xanthone that inhibits CXCR4 signaling, demonstrated that it is capable of suppressing osteoclastogenesis induced by MM cells [46][121], inhibiting activation of the NF-kB factor transcription pathway, which regulates CXCR4 expression. GA was found to suppress the SDF-1α-induced chemotaxis of MM cells and signaling pathways downstream of CXCR4 and inhibit Akt, p38, and Erk1/2 activation. Interestingly, the MM-mediated suppression of osteolytic bone damage is regulated by IL-6 inhibition and consequent p38 inhibition. These findings suggest that modulating chemotaxis factors and cytokines such as CXCR4 and IL-6, the most common factors of which include activation of the p38 signaling pathway, may be important therapeutic targets, through bifunctional alternatives or directed toward the inhibition of p38 in MM. Studies have reported that the mTOR signaling pathway is involved in the pathophysiology of MM [47][122], and a small mTOR inhibitory molecule (SC06) was found to induce the inhibition of tumor growth in an in vivo model of MM [48][123]. This inhibition did not affect other signaling pathways, such as AKT, ERK, c-Src, and JNK, but showed a significant effect on p38 in some of the analyzed MM cell lines. This suggests that the activation of the p38 signaling pathway in MM may be mediated in part by the mTOR pathway, and that the cytotoxic effect from the inhibition of the mTOR signaling pathway may be, among other pathways, the inhibition of the p38 pathway. Studies have shown that MM cell lines can express high levels of TLR5, which, when activated with its specific ligand, flagellin, induces IL-6 expression through the activation of NF-κB, which is, in turn, activated by p38 and PI3K/AKT pathway signaling. This leads to greater resistance to chemotherapeutic agents [49][124]. In addition, MM cells have also been shown to express TLR7 and TLR9 [50][125], which also induce the expression of IL-6 and, by activating the p38 pathway, induce chemoresistance [51][126]. These results suggest that there are mechanisms of the innate immune system that may favor the development and chemoresistance of MM cells, which, by activating survival signaling pathways such as p38, may play a role in the pathophysiology of MM. Thus, once again, inhibition of the p38 pathway is emerging as an important therapeutic target. All-trans retinoic acid (ATRA) has been used in the treatment of multiple myeloma, but ATRA-induced chemoresistance has been reported in myeloma patients. This is associated with the induction of apurinic endonuclease/redox factor-1 (Ape/Ref-1) expression, which leads to MDR transactivation [52][53][127,128]. Studies have revealed that ATRA activates the p38 pathway and can promote Ape/Ref-1 expression, and this was reversed by treatment with a chemical inhibitor of p38. These studies suggest that p38 has a role in chemoresistance to ATRA treatment; thus, it may be an important target in ATRA-mediated chemoresistance. Finally, as was previously discussed, treatment with arsenic trioxide (ATO) promotes p38 activation in MM cell lines, while treatment with ATO in combination with a p38 inhibitor abolished resistance to ATO, so it was suggested that p38 could be involved in the promotion of ATO chemoresistance in MM cell lines [54][70]. All these data reinforce the idea that p38 activity, especially p38 α/β, plays an important role in the pathophysiology of MM and that it directly or indirectly intervenes in the processes of chemoresistance, proliferation, and growth in MM. Additionally, it plays a role in the physiopathogenesis of MM. Therefore, its therapeutic intervention in MM continues to be a valid strategy, whether through direct inhibition or by altering any of the mechanisms that regulate its expression and activation (Figure 1).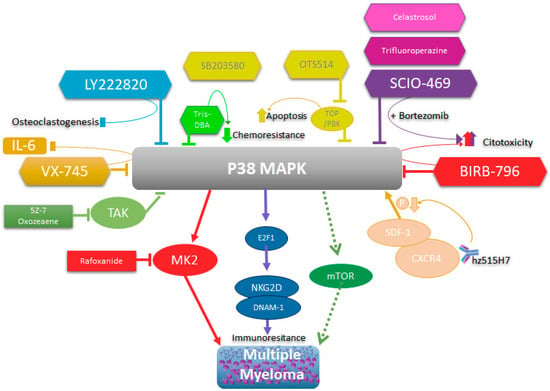
Figure 1. Schematic representation of p38’s role in multiple myeloma and its biological consequences of inhibition.