Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ashley Wang and Version 2 by Mona Zou.
The management of postoperative pain is crucial in ensuring good outcomes for surgical patients. However, results from national surveys in the United States reveal that over 80% of patients undergoing surgery complain of inadequately treated postoperative pain. Although traditional opioids such as morphine and oxycodone are commonly used in the management of acute postoperative pain, novel opioids may play a role as alternatives that provide potent pain relief while minimizing adverse effects.
- oliceridine
- tapentadol
- cebranopadol
- dinalbuphine sebacate
- nalbuphine
- dual enkephalinase inhibitors
- endomorphin-1 analog
- acute postoperative pain
1. Oliceridine
Oliceridine is a novel MOR agonist with biased ligand activity that preferentially stimulates G protein-coupling while downregulating the recruitment of β-arrestin, allowing it to limit ORAEs [1][44]. Oliceridine (brand name Olinvyk) was first approved by the FDA in 2020 to treat acute pain severe enough to warrant intravenous (IV) opioids when alternatives are inadequate [2][45]. It can be delivered in intermittent boluses or via patient-controlled analgesia (PCA). The FDA recommends administering a starting dose of 1.5 mg, with subsequent PCA demand doses of 0.35 mg to 0.5 mg with a 6 min lockout interval; the maximum total daily dose should not exceed 27 mg [2][45].
Oliceridine, an amine compound, has an onset of analgesia within 1 to 2 min, a peak effect at 6 to 12 min, and a half-life of 1.3 to 3 h [2][45]. EC50 was estimated as 10.1 ng/mL [3][46]. The chemical structure is shown in Figure 1. It is primarily metabolized in the liver by CYP3A4 and CYP2D6 P450 enzymes into inactive metabolites that are subsequently eliminated renally (70%) or fecally [2][45]. Comparing healthy subjects to those with end-stage renal disease (ESRD) and those with mild or moderate hepatic impairment, Nafziger et al. found no significant difference in oliceridine clearance. As such, no dosing adjustments are warranted for these patients [4][47]. However, they found that patients with severe hepatic impairment may benefit from reduced initial dosing and less frequent subsequent doses due to delayed clearance of the drug [2][4][45,47].
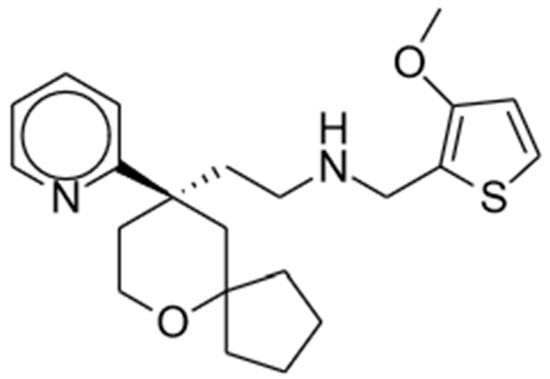
Figure 1. Oliceridine.
Oliceridine leads to adverse reactions similar to those seen in traditional opioids, including nausea, vomiting, dizziness, headache, constipation, pruritus, hypoxia, and respiratory depression [2][45]. In addition, two studies found some QTc prolongation through an unknown underlying mechanism [2][45].
1.1. Clinical Studies
In a phase I randomized, double-blind crossover study, Soergel et al. compared the safety, tolerability, and analgesia (as determined by the cold pain test) of oliceridine (1.5 mg, 3 mg, and 4.5 mg), morphine (10 mg), or placebo in 30 healthy subjects. The results showed that all three doses of oliceridine produced analgesic effects, with 3 mg and 4.5 mg having higher peak analgesia and faster onset compared to morphine. Reduction in respiratory drive was present but transient in oliceridine groups at all doses, whereas that in morphine was more persistent. In addition, severe nausea was common after morphine and oliceridine at 4.5 mg but less frequent at a 1.5 mg or 3 mg dose. Overall, the findings determined 3 mg oliceridine to have the best analgesic efficacy while minimizing the incidence of nausea and vomiting [5][30].
Viscusi et al. conducted a phase II randomized controlled trial (RCT) of 144 patients with moderate or severe acute postoperative pain after bunionectomy. Patients were given oliceridine (1, 2, or 3 mg every 3 h), morphine (4 mg every 4 h), or placebo. The results showed that 2 and 3 mg oliceridine both led to significantly lower pain intensity as assessed by a numeric rating scale (NRS) than placebo over 48 h, with meaningful pain relief within 5 min and significantly greater pain relief after the first dose compared to morphine. Furthermore, no serious adverse events were reported with oliceridine use aside from dose-related ORAEs such as nausea, vomiting, dizziness, and headaches—similar to those found with morphine use [6][31]. These findings demonstrating the rapid analgesic efficacy and tolerability of oliceridine were reflected in another phase II double-blind RCT by Singla et al. of 200 patients with moderate to severe pain after abdominoplasty. Unlike the previous study, oliceridine was administered via PCA. They found significant reduction in pain score over 24 h for oliceridine compared to placebo, providing analgesia at a similar level as morphine. The oliceridine regimen also had a faster onset and meaningful pain relief within 0.3 h compared to 1 h for the morphine regimen. Finally, there were significantly fewer nausea, vomiting, and respiratory events (e.g., hypoxia, bradypnea, hypoventilation) reported with oliceridine compared to morphine [7][48]. Both phase II studies were split into two parts with an interim analysis in between to help determine the dosing regimen for the second part [6][7][31,48].
1.2. Potential Advantages
Since oliceridine can selectively activate G-protein signaling associated with analgesia while simultaneously downregulating β-arrestin recruitment linked to ORAEs, it has potential for a widened therapeutic window. An exploratory analysis of the APOLLO studies by Beard et al. showed that when adjusted for analgesic effect, the odds of achieving “complete GI response” (i.e., no vomiting or use of rescue antiemetic) was two to three times higher with oliceridine than with morphine [8][49].
On the other hand, the APOLLO trials could not conclusively determine whether oliceridine has a lower incidence of opioid-induced respiratory depression (OIRD). Subsequently, Ayad et al. conducted an analysis of the APOLLO studies to specifically examine dosing interruption due to respiratory events and its cumulative duration as surrogate markers of OIRD. They found lower and shorter duration of dosing interruption with oliceridine compared to morphine [9][50].
Further analysis of ATHENA, which did not exclude patients at high risk of respiratory depression, also strived to investigate OIRD with oliceridine use. A retrospective chart analysis by Bergese et al. showed significantly lower incidence of OIRD events for patients receiving oliceridine compared to those on conventional opioids (8.0% vs. 30.7%) [10][51]. Future studies are warranted to better understand the association between oliceridine use and OIRD, including the ongoing phase IV VOLITION study to be completed in 2025 [1][44].
2. Tapentadol
Tapentadol (brand name Nucynta) is a centrally acting opioid analgesic that demonstrates both MOR agonist activity as well as norepinephrine (NE) reuptake inhibition [11][12][13][52,53,54]. Its chemical structure is shown in Figure 2. Tapentadol was first approved by the FDA in 2008 as an immediate-release (IR) oral tablet to treat opioid-requiring moderate to severe pain when alternatives are inadequate. Its extended-release (ER) formulation was later approved in 2011, with its use expanded for the management of neuropathic pain associated with diabetic peripheral neuropathy [11][12][13][52,53,54]. The FDA recommends starting tapentadol at a low dosage to match the treatment goals of individual patients before titrating to higher doses. The maximum total daily dose is 600 mg and 500 mg for IR and ER, respectively.
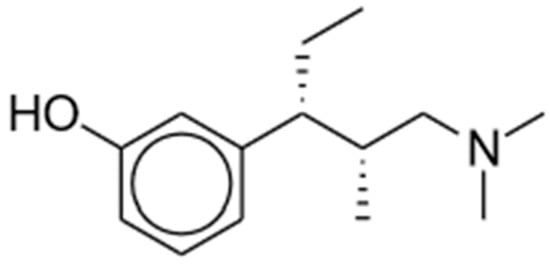
Figure 2. Tapentadol.
Tapentadol has an onset of analgesic effect of 30 min and a half-life of 4 h. It reaches peak serum concentration within an hour after administration [11][52]. EC50 was similar for both targets (1.8 μM for MOR, 2.3 μM for noradrenaline transporter) [14][55]. A total of 97% of the drug is metabolized by O-glucuronidation in the liver into inactive metabolites and subsequently excreted via the kidneys [11][52]. This is in contrast to tramadol, which requires metabolism by CYP2D6 into a potent metabolite in order to produce analgesia [15][35].
Adverse reactions commonly associated with tapentadol include nausea, dizziness, vomiting, and somnolence in adults (incidence ≥ 10%) or vomiting, constipation, nausea, pruritus, and pyrexia in pediatric patients age 6 or over (incidence > 5%). Patients taking tapentadol must be monitored closely for life-threatening respiratory depression, serotonin syndrome, adrenal insufficiency, severe hypotension, and sedation. Those at risk for adverse outcomes with tapentadol use include patients who are elderly, cachectic, or debilitated as well as those with chronic pulmonary disease or impaired consciousness. Tapentadol is contraindicated in patients with impaired pulmonary function (e.g., significant respiratory depression, acute or severe bronchial asthma), paralytic ileus, and concomitant use of monoamine oxidase inhibitors (MAOIs) or use within 14 days [11][52].
2.1. Mechanisms of Action and Preclinical Studies
A 2007 study by Tzschentke et al. examined tapentadol binding to various receptors in both rat and human cells. The results showed that tapentadol has a Ki value (indicator of binding affinity) of 0.096 μM for MOR, which is an approximately 10-fold stronger affinity compared to delta- and kappa-opioid receptors (0.97 μM and 0.91 μM, respectively) and a nearly 50 times lower affinity compared to morphine for MOR (0.0022 μM). Tapentadol binds human recombinant MOR at a similar level as rat receptors at 0.16 μM. In human transporter binding assays, the results showed tapentadol to have Ki values of 8.80 μM and 5.28 μM for NE and serotonin (5-HT) transporters, respectively, whereas no binding was determined in morphine. Despite higher affinity of tapentadol for 5-HT over NE transporters, functional assays revealed more specific and selective synaptosomal uptake inhibition of NE (0.48 μM) compared to 5-HT (2.37 μM). This suggests that stronger binding of ligands to receptors does not automatically translate to functional significance. In other words, receptor binding does not always equate to efficacy [16][56].
In addition to assessing binding affinity to receptors and functional uptake inhibition, Tzschentke et al. also studied several rat and mouse pain behavioral models to evaluate the analgesic efficacy of tapentadol, including the hot plate test, tail-flick test, mustard oil-induced visceral pain, spinal nerve ligation (SNL), and writhing response. The authors found that despite having a 50-fold lower binding affinity to MOR, tapentadol delivered analgesia at a level only 2 to 3 times lower overall compared to morphine. This hints at the key role of NE reuptake inhibition in tapentadol to relieve pain, as demonstrated in the SNL model in which the analgesic effect of tapentadol was only moderately attenuated by naloxone (a MOR antagonist) whereas the analgesic effect of morphine was completely blocked by the antagonist. Finally, using the chronic constriction injury (CCI) model of neuropathic pain, Tzschentke et al. found a delayed tolerance development to the analgesic effect of tapentadol (18 days to onset of tolerance and 51 days to complete tolerance) compared to morphine (immediate onset of tolerance and 21 days to complete tolerance) [16][56].
2.2. Clinical Studies
Tapentadol has been compared to other opioids for the management of acute pain in perioperative settings. In a systematic review of 13 studies and 1 abstract (including a total of 12,814 patients), Wang et al. examined the safety and efficacy of tapentadol IR after a variety of surgeries [15][35]; these included bunionectomy, cardiac surgery, dental procedures, total hip replacement, and abdominal hysterectomy. Results from the quantitative meta-analysis found that the lowest dose of tapentadol IR (i.e., 50 mg) was associated with less pain control, whereas higher doses (i.e., 75 mg or 100 mg) delivered a similar level of analgesia as oxycodone IR [15][17][35,57].
Qualitative synthesis by Wang et al. further examined the effect of tapentadol compared to other opioids. A phase II single-dose, double-blind RCT in 400 patients undergoing dental surgery found that higher dosage of tapentadol IR (200 mg), but not low dosage (100 mg), provided greater analgesia than morphine sulfate (60 mg) [18][58], whereas phase III multicenter, double-blind RCTs for abdominal hysterectomy (832 patients) and bunionectomy (285 patients) [17][57] showed similar levels of analgesia for tapentadol and morphine. On the other hand, comparing tapentadol and tramadol in 60 patients, Iyer et al. showed better pain control with tapentadol after cardiac surgery [19][59], while Moorthy et al. found no difference in pain score in an RCT of 100 patients experiencing acute osteoarthritic knee pain after one week of treatment [20][60]. Although it might not offer significantly improved analgesia compared with other types of opioids (e.g., oxycodone, morphine, tramadol), tapentadol appears to be better tolerated with lower incidence of gastrointestinal adverse events. Wang et al. acknowledged potential biases of the studies included in the analysis and noted that despite the variety of surgeries, the overrepresentation of minor procedures could pose a challenge in translating the results to major surgeries [15][35].
2.3. Potential Advantages
Due to its unique dual mechanism of action, tapentadol is a powerful analgesic that makes use of the synergistic interaction of two targeted receptors to address both nociceptive pain (i.e., inflammatory process from tissue damage) via MOR agonism and neuropathic pain via NE reuptake inhibition—an advantage over traditional MOR-activating opioids. Fortunately, the combined effects of the two mechanisms do not translate to a higher burden of adverse effects [21][61]. Instead, this synergy allows for less mu-opioid activity, leading to an improvement in ORAEs. This is supported by a systematic review by Freynhagen et al. in patients with moderate or severe chronic pain that found that tapentadol caused less nausea and constipation than other opioids and less dizziness and somnolence than oxycodone and oxymorphone [22][62]. Furthermore, an exploratory study by van der Schrier et al. compared the effects of tapentadol and oxycodone on the ventilatory response to hypercapnia in healthy volunteers. While tapentadol still produced respiratory depression, the 100 mg dose may have some advantage over 20 mg oxycodone, warranting further study [23][63].
Tramadol, similar to tapentadol, is an opioid analgesic with a mixed mechanism of action of weak MOR agonism as well as 5-HT and NE reuptake inhibition [13][24][54,64]. As a result, it is also considered safer than conventional opioids due to its improved adverse effect profile. However, whereas tramadol relies on metabolism by the polymorphic CYP2D6 enzyme into an active metabolite to achieve analgesia, tapentadol as a parent drug is in itself an active compound. Due to its lower serotonergic effect, tapentadol theoretically has a lower risk of causing serotonin syndrome compared to tramadol [24][64]. However, there have been reports of serotonin syndrome when tapentadol is used in conjunction with serotonergic antidepressants [25][65], though current research in the literature on the phenomenon is limited due to inadequate characterization of adverse events or lack of distinction of patients who take serotonergic medications from those who do not [26][66].
Because of its MOR agonist activity, the abuse risk of tapentadol must be carefully considered. Although the extended-release formulation of tapentadol is difficult to crush and dissolve and, thus, less likely to be misused, there have been reports of immediate-release tablets being used recreationally after being crushed and injected, at times leading to death [13][27][54,67]. Results from a study that utilized data from the National Addictions Vigilance Intervention and Prevention Program found the abuse potential of tapentadol to be higher than that of tramadol and similar to that of hydrocodone but lower than that of other strong opioids (e.g., morphine) [13][28][54,68]. Overall, tapentadol can be a safe alternative to typical opioids, especially in hospital settings and for short durations [24][64].
3. Cebranopadol
Cebranopadol is a first-in-class potent opioid receptor agonist predominantly acting at the MORs and NOP receptors with lower activity at delta and kappa receptors (MOR: Ki = 0.7 nM and EC50 = 1.2 nM; NOP: Ki = 0.9 nM and EC50 = 13 nM; delta: Ki = 18 nM and EC50 = 110 nM; kappa: Ki = 2.6 nM and EC50 = 17 nM) [29][30][69,70]. It is a spiroindole derivative of the benzenoid class and displays promising experimental results in a range of animal models and clinical trials [31][15]; the chemical structure is shown in Figure 3. Cebranopadol displays a synergistic (rather than additive) effect of the activation of NOP and classical opioid receptors [32][71].
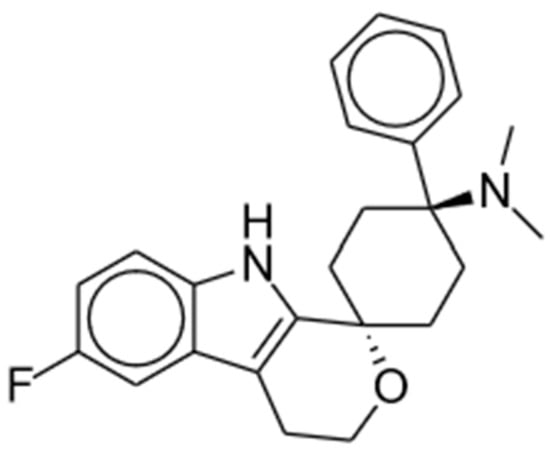
Figure 3. Cebranopadol.
Kleideiter et al. examined the pharmacokinetic characteristics of cebranopadol through noncompartmental methods in six phase I clinical trials in healthy subjects. They found that IR cebranopadol reaches maximum concentration after 4 to 6 h and has a half-value duration of 14–15 h. With once-daily dosing, the time to steady state was approximately 2 weeks, and the peak–trough fluctuation was low. Kleideiter et al. concluded that these pharmacokinetic parameters make cebranopadol a promising therapeutic option for chronic pain [33][72].
3.1. Preclinical Studies
In preclinical studies, cebranopadol has been shown to provide appropriate analgesia through peripheral and central administration in nociceptive and neuropathic pain [30][70], anti-hypersensitive effects in a rat model of arthritic pain [34][73], blockage of visceral pain [35][74], and blockage of pain in the trigeminal nerve distribution [36][75]. Additionally, Schiene et al. found that dual activation of NOP and mu receptors contributes to the anti-hypersensitive effect of cebranopadol in rat models [34][73].
There are also promising results regarding its reduced side effects and decreased use dependence. Researchers found that cebranopadol exhibits reduced respiratory depression in both adult rhesus monkeys and rats compared to fentanyl [37][38][76,77]. Through use of selective antagonists, Linz et al. mechanistically demonstrated that attenuation of respiratory depression in cebranopadol can be attributed to NOP receptor agonism which counteracts side effects resulting from MOR agonism [38][77]. Other studies indicate that cebranopadol has reduced physical dependence compared to other opioids. Tzschentke et al. conducted a direct comparison study, demonstrating that cebranopadol leads to a reduced occurrence of physical withdrawal symptoms compared to morphine [39][78]. This finding was observed across various dosages and administration periods in both mice and rats. Ruzza et al. used an elegant knockout experiment to compare symptoms of opioid withdrawal in mice. No disparity was observed in mice treated with morphine. However, cebranopadol administration induced more pronounced withdrawal symptoms in knockout mice. This finding suggests that NOP activation plays a key role in mitigating the physical dependence of cebranopadol [40][79].
Multiple studies have demonstrated that cebranopadol reduces cocaine self-administration and addiction-like behaviors in rats without directly influencing the pharmacokinetics of cocaine [41][42][43][44][80,81,82,83]. Given these results and its tolerability in humans, cebranopadol may have potential as a novel therapy for cocaine addiction. These preclinical findings highlight the potential of cebranopadol to offer reduced side effects.
3.2. Clinical Studies and Potential Advantages
Clinical studies have reaffirmed many results from animal research. In a phase I study, Dahan et al. examined the respiratory effect of 600 mcg cebranopadol on 12 healthy volunteers. Although cebranopadol produced many of the effects expected of an opioid, respiratory depression was less severe and had a ceiling due to activation at the NOP receptor. This suggests an advantage over conventional opioids, which may produce apnea at high concentrations [45][36]. In addition, another phase I study in 48 healthy nondependent recreational opioid users found a reduced peak effect of drug-liking in cebranopadol relative to hydromorphone, suggesting reduced recreational potential for the drug [46][37]. Finally, Scholz et al. conducted a phase IIa multicenter, double-blind RCT in 258 patients undergoing bunionectomy. They found that the administration of a single dose of cebranopadol at either 400 mcg or 600 mcg provided superior analgesic effects compared to 60 mg controlled-release morphine, as measured by SPI2–10 (sum of pain intensity 2–10 h after administration). While both cebranopadol and morphine produced sufficient pain relief, cebranopadol was better tolerated, and patients reported higher overall satisfaction, with the 400 mcg dose leading to less nausea, vomiting, and dizziness [47][38].
These potential advantages have also led to cebranopadol being studied for the management of chronic pain. In a phase II multicenter, double-blind RCT, Christoph et al. showed statistically significant improvements from baseline pain for all doses of cebranopadol (200, 400, or 600 mcg once daily) over placebo in 360 patients with moderate to severe chronic lower back pain that completed a 12-week period [48][39]. Similar results were seen for cancer-related pain as well. In a double-blind RCT of 126 patients, Eerdeckens et al. showed that cebranopadol at 200 to 1000 mcg was effective, safe, and well tolerated over a 7-week period. When looking at the primary endpoint of average daily rescue medication intake, cebranopadol was non-inferior and superior over prolonged-release morphine [49][84]. Furthermore, safety continues to be demonstrated when following these patients up to 26 weeks [50][85].
Overall, cebranopadol is a well-tolerated, effective analgesic that demonstrates several potential advantages over conventional opioids, including a reduced side-effect profile and lower abuse potential. According to the drug developer Tris Pharma, it is currently being tested in phase III clinical trials and has been granted fast-track status from the FDA [51][86].
4. Dinalbuphine
Dinalbuphine sebacate (DNS), a prodrug of nalbuphine, was released in Taiwan in 2017 as a result of the unmet need for long-acting analgesics. Nalbuphine is a semi-synthetic opioid that has been employed extensively in humans for years. The chemical structure is shown in Figure 4. This mu partial antagonist and potent kappa agonist has been compared to morphine and other opioids for its effective relief of moderate to severe postoperative pain [52][87]. Available under its brand name Nubain, nalbuphine has an onset of action between 2 to 3 min after IV administration and an onset of less than 15 min when administered subcutaneously or intramuscularly (IM). It has a plasma half-life of 5 h and an analgesic duration of 3 to 6 h [53][88]. The short half-life of nalbuphine indicated a need for frequent injections in clinical practice to maintain its analgesic impact.
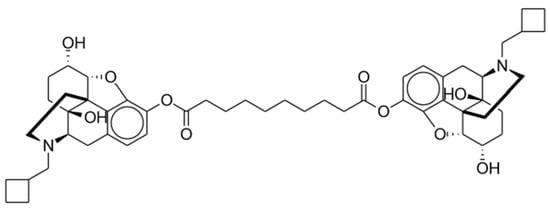
Figure 4. Dinalbuphine sebacate.
To counter the limitation of its short half-life, a single injection of 150 mg oil-based-formulation DNS allows for extended release, promising moderate to severe postoperative pain relief for 7 days [54][40]. DNS is an ester derivative of nalbuphine that requires conversion to its active component by endogenous esterases. The ester prodrug is composed of a sesame oil and benzyl benzoate solution [52][87].
5. Dual Enkephalinase Inhibitors (STR-324, PL37, PL265)
Met- and leu-enkephalins are endogenous opioid ligands that activate mu- and delta-opioid receptors (with stronger affinity for delta receptors) to produce analgesia and other effects [55][7]. Dual enkephalinase inhibitors (DENKIs), such as STR-324, PL37, and PL265, work by blocking both of the two major enkephalin-degrading enzymes (neprilysin and aminopeptidase N). The chemical structures of DENKIs are shown in Figure 5. This inhibition results in higher synaptic concentrations of the signaling molecules and increased stimulation of opioid receptors at both central and peripheral receptors [56][93]. DENKIs have been shown to produce analgesic effects in animal models and preliminary human studies with fewer adverse effects than conventional opioids [57][94].
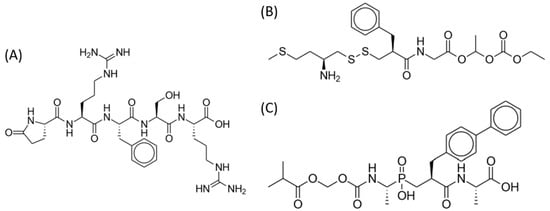
Figure 5.
Dual enkephalinase inhibitors. (
A
) STR-324. (
B
) PL37. (
C
) PL265.
There are certain disadvantages associated with exogenous opioids. Drugs administered externally do not target specific locations or achieve the same concentrations as naturally occurring ligands within the body. Additionally, the levels of endogenous neurotransmitters in the synaptic system are closely regulated through homeostasis. As such, external ligands may disturb normal control mechanisms [57][94]. Theoretically, inhibition of the degradation of endogenous opioid ligands can avoid some of the shortfalls of exogenous opioids and produce a more natural analgesic effect.
5.1. Preclinical Studies
Despite the limited number of human studies conducted on DENKIs, preclinical investigations show encouraging outcomes suggesting that DENKIs PL37, PL265, and STR-324 may provide efficient pain relief while minimizing specific side effects typically linked to conventional opioids.
Preclinical research on the DENKI PL37 has shown promising results in various models of pain and migraine. Studies conducted on mice and rats demonstrated that PL37 administration, either IV or orally (PO), effectively attenuated stress-induced periorbital hypersensitivity, facial grimace responses, and cutaneous hypersensitivity [58][59][95,96]. Furthermore, PL37 exhibited antinociceptive and anti-allodynic effects in a rat model of peripheral neuropathic pain [59][96]. It also suppressed osteosarcoma-induced thermal hyperalgesia in mice when administered PO [56][93]. Menendez et al. concluded that PL37 activated micro-opioid receptors because administration of cyprodime, a specific antagonist of the micro-opioid receptor, inhibited antihyperalgesic effects [56][93].
Topical instillations of PL265 significantly decreased corneal inflammation in a corneal inflammatory pain model [60][97]. Additionally, PL265 demonstrated preventive and alleviative effects in a murine model of neuropathic pain, acting specifically at the level of peripheral nociceptors. The repeated administration of PL265 did not induce tolerance, making it a promising approach to prevent and alleviate neuropathic pain without the unwanted effects of traditional opiates [61][98]. Furthermore, PL265 exhibited long-lasting oral analgesic effects, and its efficacy was mediated through stimulation of peripheral opioid receptors as demonstrated by naloxone methiodide reversion [62][99].
In a rat model of mononeuropathy, continuous IV administration of STR-324 over seven days significantly reduced pain-related behavior, along with the pain-evoked expression of spinal c-Fos, demonstrating that the drug acts at least in part through inhibition of endogenous nociceptive pathways [63][100]. STR-324 infusion also resulted in reduced responses to mechanical stimuli, with its antinociceptive effect being reversed by naloxone, an opioid antagonist [64][101]. Notably, during the three-day postoperative period, no adverse effects on respiratory rate, oxygen saturation, arterial pressure, or heart rate were induced by opiorphin [64][101].
5.2. Clinical Studies
The only published DENKI trial in humans to date is a randomized, double-blind, placebo-controlled ascending dosing study in 78 volunteers to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of STR-324 [65][43]. Data from the study suggest a favorable safety and tolerability profile in healthy males up to a dose of 11.475 mg per hour administered through a 48 h infusion. While some adverse effects occurred during the course of the experiment, no dose-dependent effect was observed in relation to STR-324. The exact pharmacokinetic parameters of STR-324 were not determined given the difficulty in measuring quantifiable concentrations in the plasma. However, the team did conclude that STR-324 undergoes rapid metabolism and quickly distributes beyond the bloodstream after administration.
However, no consistent antinociceptive action of STR-324 was observed during the study. Moss et al. postulate that this may be due to decreased involvement of endogenous enkephalinases in the acute pain models used. It is possible that the PainCart-evoked pain model does not create sufficient pain intensity for an effect of STR-324 to be shown or that enkephalinases are only significantly active in prolonged pain [65][43].
DENKIs have shown promising preclinical results with regards to anti-nociception, tolerability, and reduced side-effect profiles compared to conventional opioids. Further clinical research is necessary to determine the effectiveness, dosing, pharmacokinetic parameters, and side-effect profiles of DENKIs in humans. To date, several clinical trials have been registered by pharmaceutical companies including PL37 (phases I and II) and PL265 (phase II) by Pharmaleads as well as STR-324 in phase II by Stragen [66][67][68][102,103,104]. These studies indicate that DENKIs are generally well tolerated and safe in humans through PO administration.