Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by LAURA CONTI and Version 2 by Lindsay Dong.
TLR2 activation by pathogen-associated or damage-associated molecular patterns (PAMPs or DAMPs, respectively), activates a signaling cascade initiated by Myeloid Differentiation Primary Response protein 88 (MyD88). The consequent recruitment of the interleukin-1 receptor-associated kinase (IRAK)–TNF Receptor Associated Factor (TRAF) complex leads to the activation of NF-κB and MAPK pathways. This induces the production of pro-inflammatory cytokines, and in cancer cells, may stimulate epithelial to mesenchymal transition (EMT) and proliferation.
- breast cancer
- Toll-like receptor 2
- TLR2
1. Introduction
Breast cancer is the most prevalent malignancy in women and ranks as the second leading cause of cancer-related deaths on a global scale [1]. Despite advancements in the treatment of breast cancer, a significant proportion of patients still advance to metastatic disease, posing a considerable challenge to effective therapy. The limited success of current therapies often stems from a predominant focus on targeting cancer cells alone, overlooking the intricate interactions within the tumor microenvironment (TME). Various cell populations in the TME engage in complex communication with cancer cells, facilitating cancer progression and resistance to treatments. In some instances, the complex crosstalk between cancer and immune cells leads to immunosuppression, redirecting the immune system toward a protumorigenic response.
Dysregulation of innate immunity is often associated with breast cancer and significantly contributes to inducing a chronic inflammatory state within the TME—a hallmark of cancer that contributes to all steps of cancer development and to resistance to current therapies [2]. Moreover, some of the receptors typically expressed by innate immune cells, such as pattern recognition receptors (PRRs), may promote tumor growth through intrinsic mechanisms within cancer cells [3].
Mounting evidence suggests a significant association between Toll-like receptors (TLRs) and the development of breast cancer [4][5][4,5]. TLRs exhibit a dual role in cancer: they can mediate tumor cell death, activating an effective antitumor immune response, or inhibit the immunosuppressive activity of myeloid-derived suppressor cells (MDSCs), preventing tumor growth [6][7][6,7]. However, they also possess protumorigenic functions [7]. This is particularly evident for TLR2, the most expressed TLR in triple negative breast cancer [4]. Indeed, data from the Kaplan–Meier plotter (kmplot.com (accessed on 21 December 2023)) and from the literature indicate that high expression of TLR2 is associated with poor relapse-free survival in breast cancer patients [8]. On the contrary, the expression levels of the other TLRs are not associated with poor prognosis in breast cancer (kmplot.com (accessed on 21 December 2023)). TLR2 activation by pathogen-associated or damage-associated molecular patterns (PAMPs or DAMPs, respectively), activates a signaling cascade initiated by Myeloid Differentiation Primary Response protein 88 (MyD88). The consequent recruitment of the interleukin-1 receptor-associated kinase (IRAK)–TNF Receptor Associated Factor (TRAF) complex leads to the activation of NF-κB and MAPK pathways. This induces the production of pro-inflammatory cytokines, and in cancer cells, may stimulate epithelial to mesenchymal transition (EMT) and proliferation (Figure 1). TLR2 promotes cancer cell survival and proliferation in breast [8] and gastric cancers [9], as well as in pancreatic ductal adenocarcinoma [10]. However, some studies suggested that TLR2 activation by PAMPs, or DAMPs such as high-mobility group box 1 (HMGB1) and heat shock proteins (HSPs), may promote anticancer immune responses [11][12][13][11,12,13]. The conflicting reports on TLR2’s antitumor and protumor properties underscore the critical need to comprehend its context-dependent role in breast cancer for potential therapeutic advancements.
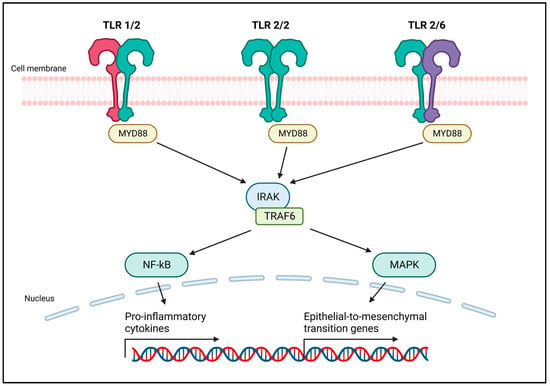
Figure 1. Schematic representation of TLR2 dimers and their signaling pathway. Like other TLRs, TLR2 forms homo- or heterodimers that allow for activation and signaling upon ligand binding. TLR2 islocalized in the outer cell membrane, and mainly dimerizes with TLR1 and TLR6. TLR2 uses the canonical MyD88 pathway to transduce a signal that, through the IRAK–TRAF6 complex, induces the activation of NF-κB and MAPK. NF-κB is responsible for the transcription of several pro-inflammatory cytokines. The MAPK pathway induces the epithelial to mesenchymal transition, promoting cancer cell invasion and metastasis. Created with BioRender.com (accessed on 22 December 2023).
2. Role of TLR2 in Anticancer Immune Responses
TLRs are fundamental tools exploited by the immune system to trigger the innate immune response against pathogens and subsequently activate adaptive immunity. The immune surveillance is not only focused on preventing or fighting infections. It is also crucial for identifying and eliminating malignant cells that can initiate tumor development. When a tumor arises, the immune system engages in a complex process to counteract tumor progression, and attempts to resist cancer-induced immune suppression. In this context, the activation of TLRs by DAMPs released from cancer cells may enhance antigen presentation mechanisms in dendritic cells (DCs) and promote macrophage differentiation towards the M1 phenotype, known for its antitumor activity [14][20]. Consequently, TLR2 agonists have been evaluated in several solid tumor models as adjuvants for immune stimulation. For instance, the TLR2 natural ligand polysaccharide krestin (PSK) was able to stimulate TLR2 and elicit NK cell-mediated antitumor responses in different cancer models. In particular, PSK potentiated trastuzumab-induced antibody-dependent cell cytotoxicity of HER2+ breast cancer cells by stimulating NK cell activation [15][21]. To confirm that TLR2 stimulates NK cells, Ke et al. demonstrated that treatment with Strongylocentrotus nudus egg polysaccharide, a TLR2 agonist, induces NK cell proliferation, cytotoxicity and release of interleukin (IL)-2 and IFN-γ in a mouse model of lung cancer [16][22]. In lung cancer preclinical models, the administration of PAMPs or synthetic TLR2 ligands induced the differentiation of M1 macrophages that release nitric oxide, IFN-γ and pro-inflammatory cytokines, suggesting that TLR2 activation favors antitumor immune reactions [17][23]. In melanoma, TLR2 stimulation using the synthetic compound diprovocim was reported to induce antitumor activity in response to ovalbumin vaccination [18][24].
Thus, the role of TLR2 in anticancer immune responses remains controversial and largely reliant on tumor types and models. TLR2 appears to possess the potential to either stimulate the immune system or suppress it. Part of this controversy might arise because TLR2 expression is not confined to the immune system. Many cancer cell types express TLR2 and take advantage on the activation of its signaling pathway, as will be discussed in the following paragraph.
3. The Protumoral Role of TLR2 in Breast Cancer
Immune cells exploit PRRs to detect disruptions in tissue homeostasis caused by cancer cells, thereby triggering an antitumor immune response. However, cancer cells can express PRRs as well, benefiting from the activation of their signaling pathways [19][26]. Among the PRRs, members of the TLR family exhibit an intriguing dual role in cancer. TLR2 mRNA expression is significantly higher in breast cancer than in normal tissues, with a higher expression in the triple-negative and HER2+ subtypes as compared to the luminal A and luminal B [8]. A significant increase in the levels of soluble TLR2 was observed in the sera from patients with both metastatic and non-metastatic breast cancer, as compared with healthy donors. Of note, soluble TLR2 was significantly higher in metastatic than in non-metastatic breast cancer patients, suggesting that TLR2 might be used as a biomarker to monitor disease progression [20][27]. Moreover, a significant correlation was observed between high TLR2 expression and poor prognosis in breast cancer patients [8]. Similarly, a positive correlation exists between the expression levels of its partners, TLR1 and TLR6, and the development of brain metastases in breast cancer patients [21][28], indicating the potential involvement of the TLR2 heterodimers in the metastatic spread of pre-existing tumors. Furthermore, high TLR2 mRNA levels are associated with poor relapse-free and overall survival in breast cancer patients who underwent surgery [8], as well as in patients treated with endocrine therapy or chemotherapy [8][22][8,29]. Importantly, TLR2 expression levels can predict the response to endocrine therapy with high accuracy in both luminal A and luminal B breast cancer patients [8]. Resistance to both endocrine therapy and chemotherapy has been associated with the presence of cancer stem cells (CSCs) [23][30]. TLR2 is expressed by breast cancer cells and promotes CSC self-renewal, invasiveness and drug resistance [24][31]. These effects result from various mechanisms, mostly dependent on the availability of TLR2 ligands, particularly DAMPs, in the TME. These molecules can be actively secreted by cancer cells or passively released during chemo- or radiotherapy, activating TLR2 signaling in either cancer or immune cells. Among the DAMPs that induce TLR2 activation, HMGB1 plays a major role. In the nucleus, HMGB1 is involved in the DNA repair processes and transcription, interacting with transcription factors like p53 [25][32]. However, HMGB1, as other moonlighting proteins, not only acts as a DNA-binding protein but also functions outside the cell. It can be actively secreted in response to cytokine stimulation or passively released from necrotic or damaged cells, subsequently inducing TLR2 activation [26][33]. Notably, breast cancer patients exhibiting high HMGB1 expression are more prone to develop cancer metastasis, especially in triple-negative breast cancer [27][34]. Other important DAMPs activating TLR2 include heat shock proteins (HSPs), a family of proteins that play a dual role. While their intracellular function involves supporting the correct folding or refolding of nascent and misfolded proteins, they can also be actively secreted or released by necrotic cells, binding to TLR2 in the TME. In breast cancer, the extracellular HSP90 co-chaperone Morgana acts as another significant activator of TLR2 [28][35], whereas versican has been identified as the DAMP involved in TLR2-mediated tumor promotion in glioma and lung cancer [29][36]. TLR2 serves also as a sensor of PAMPs, representing a bridge between eukaryotic cells and the microbial world, whose alterations may influence cancer development in different ways. In addition, TLR2 expressed on immune cells may not only exert antitumor activity but also lead to several immune suppressive effects that indirectly promote cancer progression [30][37].3.1. The Cancer Cell-Intrinsic Protumoral Effects of TLR2
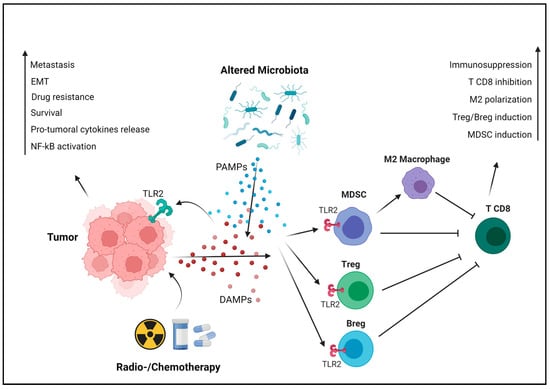
In breast cancer cells, TLR2 is overexpressed and can be activated by endogenous ligands such as HSPs, HMGB1 and other DAMPs, or by exogenous ligands derived from pathogens, like bacterial lipoproteins. Once activated, TLR2 signaling promotes tumor growth and survival. Among the critical downstream molecules activated by TLR2 is NF-κB, which triggers the transcription of various pro-survival and pro-inflammatory genes [30][37]. Wenjie Xie and colleagues have demonstrated that TLR2 activation promotes breast cancer cell survival, proliferation and invasion through the activation of NF-κB and the secretion of protumoral cytokines. They also reported a 10-fold higher TLR2 expression in the invasive MDA-MB-231 human triple-negative breast cancer cells compared to the poorly invasive ER+ MCF-7 cells [31][38]. TLR2 plays a role in the regulation of CSCs, a subpopulation of tumor cells with self-renewal and tumor-initiating capabilities. Activation of TLR2 in mammary epithelial stem cells and in breast cancer cells enhances the expression of stemness-related genes, promoting the acquisition of CSC properties that contribute to treatment resistance and tumor recurrence [32][39]. Apart from its direct effect on tumorigenesis, scholars have demonstrated that TLR2 mediates breast cancer resistance to doxorubicin and other drugs, inducing immunogenic cell death (ICD) and the release of DAMPs that activate TLR2 signaling in cancer cells, thereby enhancing their survival [22][29]. In addition to NF-κB, TLR2 signaling activates the MAPK pathway, involved in cell proliferation and migration. In gastric cancer, this activation leads to the EMT, a pivotal process linked to cancer cell invasion and metastasis. TLR2-mediated EMT allows cancer cells to acquire a more migratory and invasive phenotype, facilitating their dissemination to distant sites and the formation of metastasis (Figure 2) [33][41]. Another important aspect of TLR2’s protumoral role is its impact on angiogenesis, the process of forming new blood vessels to supply nutrients and oxygen to growing tumors. TLR2 activation in breast cancer cells upregulates the expression of vascular endothelial growth factor (VEGF) and other pro-angiogenic factors, fostering the formation of new blood vessels that support tumor growth [31][38].Figure 2. TLR2-mediated protumoral mechanisms. Cancer cells actively or passively release DAMPs, especially following cell death induced by chemo- or radiotherapy. In addition, alterations in the microbiota due to antibiotic therapies, chemotherapies, or other factors can foster the proliferation of bacterial species that exert protumoral activity, releasing PAMPs in the TME. These endogenous and exogenous TLR2 ligands activate its signaling, promoting tumor progression in a cancer cell-intrinsic manner (on the left). TLR2 signaling activates NF-κB, which subsequently transcribes protumoral cytokines such as IL-6, TGF-β and VEGF. This stimulates cancer cell survival, angiogenesis and resistance to therapies. Additionally, TLR2 triggers the MAPK pathway, inducing EMT and promoting metastasis. Moreover, TLR2 is expressed by immune cells, where it exerts immunosuppressive effects favoring tumor progression in a cancer cell-extrinsic manner (on the right). TLR2 induces the differentiation of T and B regulatory cells, as well as of MDSCs from bone marrow precursors. MDSCs contribute to immunosuppression through several mechanisms, including the reprogramming of macrophages into the M2 phenotype. These processes collectively result in the production of cytokines that inhibit CD8+ T cells and their activity, promoting tumor immune evasion. Created with BioRender.com (accessed on 10 November 2023).
Beyond its direct effects on cancer cells, TLR2 may also confer protection from CD8+ T cell killing. Other TLRs, such as TLR4, induce the expression of immune checkpoint molecules like programmed death-ligand 1 (PD-L1), which can lead to T-cell exhaustion and suppress antitumor immune responses [34][42]. However, the correlation between TLR2 and PD-L1 expression requires further elucidation.
TLR2 serves as both a DAMP detector and a sensor of PAMPs, connecting TLR2-expressing cells with the microbial world. This mechanism has been largely studied in the immune system for its protective role against infections. Recently, researchers have focused on studying the role of the microbiota in different diseases. Alterations in the microbiota composition, and its interaction with our cells, might be involved in many pathogenic processes, including carcinogenesis and cancer recurrence [30][35][37,43]. Several studies have demonstrated the correlation between specific bacterial species and tumor development, especially in the gastrointestinal system. For instance, Helicobacter pylori contributes to gastric and colon carcinogenesis through various mechanisms, including TLR2-mediated activation of NF-κB and Wnt, and the induction of EMT in epithelial cells [36][37][44,45].
A specific microbiota has been detected in the mammary gland, altered in breast cancer. Bacteroides fragilis is found in tumor biopsies of breast cancer patients, and promotes tumorigenesis. Fusobacterium nucleatum (F. nucleatum), an oral commensal bacterium, can spread to other organs and become pathogenic. Indeed, F. nucleatum is detected in colon and breast cancer tissues, directly promoting tumor progression by activating TLR2 signaling in cancer cells and inducing immunosuppression, as described in the following paragraph [38][39][40][46,47,48].
Beyond the individual protumoral role played by specific bacterial species under certain conditions, it is important to note more complex alterations in the microbiota across various parts of the body. These conditions, called dysbiosis, can cause carcinogenesis or promote the progression of existing tumors by enhancing resistance to therapies and the spread of metastasis. Correlations between antibiotic therapies before cancer diagnosis or in its early stages and poorer prognosis in breast cancer patients have been reported. This is probably caused by the establishment of dysbiosis and increased presence of protumoral bacteria. Furthermore, chemotherapy can induce dysbiosis and opportunistic infections, increasing the availability of TLR2 ligands and potentially limiting therapy effectiveness [41][42][43][49,50,51].
Collectively, this evidence strongly suggests that in breast cancer, the presence of TLR2 along with its endogenous or exogenous ligands can significantly influence a worse prognosis.
3.2. The Cancer Cell-Extrinsic Protumoral Effects of TLR2
TLR2 does not just impact cancer cells directly; it also plays a significant role in cancer progression through its influence on the immune system. When activated, TLR2 can either promote or hinder tumor growth, depending on the specific immune cells involved. Tregs, MDSCs, neutrophils and macrophages express TLR2, and upon its activation, they contribute to cancer progression and metastasis due to their immunosuppressive functions [30][37]. Treg frequency is significantly decreased both in the TME and in the periphery in TLRKO breast tumor-bearing mice, since TLR2 activation on Treg cells induces their expansion [22][44][29,52]. Moreover, upon TLR2 stimulation, macrophages release chemokines that recruit Treg [45][53]. Tregs subsequently inhibit the antitumor activity CD8+ T cells through the release of immunosuppressive cytokines, like IL-10 [30][37]. Similarly, TLR2 activation in B lymphocytes induces their differentiation into regulatory B cells (Bregs) that produce IL-10 and suppress the T-cell antitumor response [46][54]. TLR2 activation in CD4+ T cells, triggered by HSP90 on autophagosomes released by breast cancer cells, initiates an autocrine IL-6 cascade. This process induces the expression of IL-10 and IL-21, fostering immune suppression and inhibiting antitumor responses [47][55]. TLR2 activation in bone marrow precursors induces their differentiation to MDSCs, which accumulate in the TME and release protumoral cytokines. Moreover, this contributes to the polarization of macrophages towards the protumoral M2 phenotype and the release of nitric oxide (NO), a potent inhibitor of effector T cells. DAMPs such as HMGB1 and serum amyloid A 1 protein, secreted by breast cancer cells and elevated in the plasma and tumor biopsies from patients with advanced triple-negative breast tumors, induce an immunosuppressive response in neutrophils via TLR2 [27][48][34,56]. Specifically, HMGB1 prompts the release of neutrophils’ extracellular traps, favoring the development of lung metastasis in triple-negative breast cancer mouse models [27][34].
These mechanisms collectively create an immunosuppressive microenvironment that supports tumor progression. TLR2 activation within immune cells triggers the release of various factors that hinder immune responses against tumors, promoting their growth and spread. Understanding these interactions helps identify potential targets to disrupt the immunosuppressive TME and bolster antitumor immunity.