Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jason Zhu and Version 1 by Davide Mauri.
Gastric cancer (GC) is one of the most common and aggressive types of cancer. Immune checkpoint inhibitors (ICIs) have proven effective in treating various types of cancer. The use of ICIs in GC patients is currently an area of ongoing research. The tumor microenvironment (TME) also seems to play a crucial role in cancer progression. Tumor-associated macrophages (TAMs) are the most abundant population in the TME. TAMs are capable of displaying programmed cell death protein 1 (PD-1) on their surface and can form a ligand with programmed death ligand 1 (PD-L1), which is found on the surface of cancer cells. Therefore, it is expected that TAMs may significantly influence the immune response related to immune checkpoint inhibitors (ICIs).
- tumor microenvironment (TME)
- tumor-associated macrophages (TAMs)
- immune checkpoint inhibitors (ICIs)
- PD-1/PD-L1
- gastric cancer
1. Introduction
Gastric cancer (GC) is one of the most common and aggressive types of cancer. It has the fifth-highest incidence of all cancers and is presently the fourth leading cause of cancer death worldwide [1]. Due to their extensive morphological variability, GC can be classified through various approaches. The widely used tumor–node–metastasis (TNM) classification stratifies it into superficial (T1) or advanced types (T2–T4). Morphological distinctions can also be made based on characteristics such as the presence of mucus in the cytoplasm and the rate of glandular vs. tubular differentiation. Although all gastric adenocarcinomas originate from the stomach’s glandular epithelium, their morphology varies based on the location of affected glands (pyloric, fundic, cardiac), the extent of surface involvement, and potential metastatic lesions [1,2,3,4][1][2][3][4].
Moreover, histologically, GC has different subtypes, and for this reason, there is a classification method known as Lauren’s criteria that has implications for therapy. The subtypes are as follows: intestinal, diffuse, and mixed types. Notably, diffuse-type adenocarcinoma tends to be more frequently diagnosed in female and younger patients, while the intestinal type is often associated with Helicobacter pylori infection and the presence of intestinal metaplasia [4,5,6][4][5][6]. Figure 1, shows the histological image of GC cells. Chemotherapy remains the main treatment option for metastatic GC; however, due to the tumor’s heterogeneity, the patients’ prognosis is poor. The emergence of molecular profiling of tumors in clinical settings paves the way for the application of novel targeted therapies with chemotherapy [4,5][4][5].
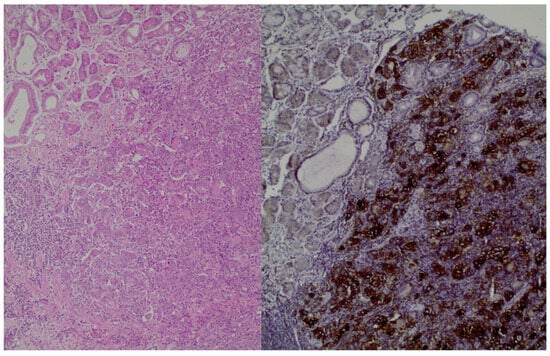
Figure 1. (left): Gastric adenocarcinoma, Hematoxylin and eosin (H+E) × 200, (right): Gastric adenocarcinoma, PD-L1 immunostaining (DAKO), DAB × 200.
For instance, trastuzumab, a monoclonal antibody targeting human epidermal receptor 2 (HER2), was the first approved targeted therapy. Unfortunately, only 15–20% of patients can benefit from trastuzumab. As a second-line therapy, another targeted therapy, anti-vascular endothelial growth factor therapy was approved [4,5,6,7][4][5][6][7]. Current research is focused on improving GC treatment outcomes by identifying new therapeutic targets related to proteins involved in epithelial–mesenchymal transition (EMT) and cell–cell adhesion. EMT plays a vital role in cancer metastasis and is initiated by the breakdown of cell–cell adhesion structures such as tight junctions, adherens junctions, desmosomes, and gap junctions. Among these, claudins (CLDNs) exhibit high expression levels in certain cancers, including GC. The effectiveness of these approaches in an adjuvant setting is still being explored [8,9][8][9]. Aberrations in CLDN18.2, found predominantly in the genomically stable subgroup and diffuse histological subtype, represent promising targets for precision drugs like monoclonal antibodies. Zolbetuximab, a monoclonal antibody designed specifically for CLDN18.2, has exhibited effectiveness in phase II trials and, more recently, in the phase III SPOTLIGHT trial. The results demonstrated improved progression-free survival (PFS) and overall survival (OS) compared to standard treatment protocols. Moreover, the phase III GLOW trial demonstrated that zolbetuximab, when used in combination with chemotherapy, improved both PFS and OS in patients with CLDN18.2-positive, HER2-negative, locally advanced unresectable or metastatic GC [9,10,11][9][10][11]. Fibroblast growth factor receptor 2 (FGFR2), a member of the fibroblast growth factor family, is active in about 20–30% of GCs. This activation is primarily attributed to the amplification of the FGFR2 gene or the presence of splice variants. Bemarituzumab has the potential to target this activation in GC. To assess bemarituzumab’s effectiveness, the FIGHT study selected patients who showed FGFR2b membrane overexpression in at least 5% of tumor cells via immunohistochemistry (IHC) [4]. Additionally, there has been significant emphasis in the scientific community on using immune checkpoint inhibitors (ICIs) to prolong patient survival. The ICIs are specialized monoclonal antibodies that focus on disrupting the cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and PD-1/PD-L1 pathway. The use of the anti-CTLA-4 inhibitor (Ipilimumab) was first established in the treatment of melanoma [12,13,14,15][12][13][14][15]. Currently, a wide spectrum of cancer types can be effectively addressed through anti-PD-1 agents (such as pembrolizumab and nivolumab), anti-PD-L1 inhibitors (like avelumab and atezolizumab), or through a co-administration of these agents with an anti-CTLA4 inhibitor [12,13,14,15,16][12][13][14][15][16].
Consequently, incorporating anti-programmed cell death protein 1 (PD-1) into first-line treatments for HER-2 negative GC improves clinical outcomes only in patients with high programmed death ligand 1 (PD-L1) expression. It should be emphasized that in the phase II randomized CheckMate 649 trial, it was found that the combination of nivolumab and chemotherapy is a recommended first-line treatment for patients with HER2-negative GC, especially those with a PD-L1 expression level of 5 or more, as assessed by the combined positive score (CPS) [11]. The Keynote-059 study (a phase II study) also showed that pembrolizumab plus chemotherapy demonstrates manageable safety and promising antitumor activity as first-line therapy in advanced GC, regardless of PD-L1 expression [12]. Additionally, the Keynote-811 study showed that pembrolizumab, in combination with trastuzumab and chemotherapy, significantly improved PFS in patients with metastatic HER2-positive GC, specifically with a PD-L1 CPS of 1 or more [14]. Moreover, patients who express high microsatellite instability seem to experience impressive clinical improvement and prolong survival.
Furthermore, although there have been significant advances in diagnostic techniques and treatment methods, GC remains associated with a poor prognosis. Therefore, it is essential to investigate the immunomodulatory role of the tumor microenvironment (TME) and its influence on the effectiveness of ICIs. This is important crucial for the improvement of currently approved therapies [16].
The principal role of immune checkpoint blockade is to counteract the suppression of T cells caused by the tumor, effectively “reawakening” them. Consequently, the immune system is able to identify and eliminate the tumor through a series of orchestrated mechanisms [10,11,12,13,14][10][11][12][13][14].To be more specific, PD-1 is a 55-kDa transmembrane glycoprotein and contains an extracellular immunoglobulin variable (IgV)-like domain responsible for connecting with its ligands. Additionally, it has a cytoplasmic tail containing two tyrosine-based inhibitory motifs (ITIMs), the role of which is to suppress the immune system. Specifically, PD-1 and its ligands (PD-L1/PD-L2) are involved in the self-recognition and protection process, inhibiting the function of immune cells [14,15,16][14][15][16]. For this reason, a cancer cell may be recognized as a potential harm and targeted by the immune system; however, when interacting with the PD-1/PD-L1/PD-L2 signaling, it is recognized as part of the self and, therefore, rescued from the immune system targeting (cancer escape). The blockade of the PD-1 axis bypasses self-immuno-recognition and has been shown to be an effective treatment in several types of cancer. For this reason, many clinical trials have examined the benefit of PD-1/PD-L1 inhibitors in GC [12,13,17,18,19,20][12][13][17][18][19][20].
2. Correlation of TAMs and PD-L1
There is a correlation between the presence of TAMs and the expression of PD-L1. GC tissues with high PD-L1 expression show significantly greater macrophage infiltration. Conversely, low PD-L1 expression in GC is associated with reduced infiltration of TAMs. Whether these small differences in TAM expression may play a crucial role in immunotargeting/immunoescaping and whether they might influence immunotherapy outcomes remains a subject of research, as no prospective randomized clinical data are currently available [39,40,41,42,43,44,45][21][22][23][24][25][26][27]. Specifically, in Ju et al.’s study, it is reported that if there is no expression of PD-L1 in the cancer cells, the infiltration of CD68 cells is only 42%. On the other hand, if there is an expression of PD-L1, the infiltration of CD68 cells is estimated at 58%. Therefore, there is a positive correlation between the expression of PD-L1 and the infiltration of CD68 cells. Additionally, it is suggested that differentiated macrophages can induce PD-L1 expression in GC cells [40][22]. The study conducted by Junttila et al. revealed that 41% of the patients with GC had a high density of clever-1-positive macrophage, and 48% of them exhibited a high immune cell score. Additionally, it was noted that PD-L1 CPS (p = 0.474) was not statistically associated with survival. Similar results were observed between low and high densities of PD-1-positive lymphocytes (p = 0.204) and clever-1-positive macrophages (p = 0.428). Moreover, in the high ICS group, patients with high PD-L1, PD-1 or clever-1 had poor prognoses [41][23]. In addition, Huang et al.’s study shows that all TAMs express PD-L1. Notably, the macrophages characterized by the CD68+ CD206+ phenotype exhibit a substantially higher average expression of PD-L1 per patient when compared to other TAM subsets. The study also noted the heterogeneity of macrophages within the tumor, attributing it to the various markers used for their characterization [42][24]. Moreover, Harada et al.’s study shows that the density of CD68- and CD163-positive cells in PD-L1-positive GC samples was significantly higher than in PD-L1 negative GC samples (CD68 p = 0.0002; CD163 p < 0.0001). The study concludes that M2 macrophage infiltration could serve as a predictive marker for PD-L1 expression, thereby making M2 macrophages a potential therapeutic target in GC [43][25]. Finally, Ivanovi et al.’s study identified a link between the presence of PD-L1 expression in tumor cells and the infiltration of macrophages. To be more precise, the proportion of macrophages within PD-L1-positive GC instances was found to be 2.6 times greater compared to the levels observed in PD-L1 negative cases [45][27]. Ubukata et al.’s study is the only study that did not find a statistically significant correlation between PD-L1 expression and CD163 (p-value = 0.6) [39][21]. It is believed that TAMs are recruited from both tissue-specific embryonic and monocytic-derived resident macrophages, with the latter source contributing to the growing body of TAMs. Macrophages can be differentiated by elevated levels of different types of cytokines, chemokines, growth factors and other signals from tumor and stromal cells. M1 macrophages secrete proinflammatory cytokines such as IL-12, tumor necrosis factor (TNF-α), chemokine ligand 1 (CXCL-10), and interferon (IFN)-γ and produce high levels of nitric oxide synthase (NOS), which is responsible for the metabolizing arginine to the nitric oxide. On the other hand, M2 macrophages secrete anti-inflammatory cytokines such as IL-4, IL-10, and IL-13 and express in high degrees arginase-1, CD206, and CD163 [33,39,40,41,42,43,44,45][21][22][23][24][25][26][27][28]. It seems that IL-6 and TNF-α are responsible for the high expression of PD-L1. The NF-κβ and STAT3 signaling pathways regulate macrophage-induced PD-L1 expression. STAT3 inhibitor C188-9 and IKK inhibitor BAY11-7821 can suppress the expression of PD-L1 [21][29]. In addition, it seems that TNF-α, p-65, and STAT3 have a good prognostic value in GC, and macrophages can promote the proliferation of GC cells by inducing the expression of PD-L1 [33,40][22][28]. In addition, the infiltration of TAMs is closely associated with tumor invasion and the development of metastasis. In this context, tumor cells release colony-stimulating factor 1, while TAMs release epithelial growth factor (EGF). These factors collectively promote the co-migration and invasion of both cell types toward blood vessels. It has been observed that macrophages from distant metastatic sites enhance the rate of invasion when compared to non-metastatic cells. Notably, the migration rate experiences a significant boost when a cell line containing macrophages is cultured under hypoxic conditions. In response to hypoxia, there is an upregulation in the expression of genes encoding disintegrin and metalloproteinase domain-containing protein 8 and 9 (ADAM8 and ADAM9), while the expression of matrix metallopeptidase 9 (MMP9) and tissue inhibitor of metalloproteinase 3 (TIMP3) decreases. This modulation in the expression of ADAMs, TIMP3, and MMP9 suggests that these genes may collectively contribute to the role of TAMs in facilitating the rapid and aggressive invasion characteristic of GC [3].3. Prognosis of PD-1+ TAMs
Wang et al.’ s study found that the macrophages in tumor tissues express a higher level of PD-1 compared to those in non-tumor tissues and in blood samples. PD-1+ TAMs were also accumulated in GC tissue [32,33,34][28][30][31]. In Kono et al. ’s study, the correlation between the presence of PD-1+ TAMs and clinicopathological variables is examined. High PD-1+ TAMs is defined as the frequency of PD-1+ macrophages: ≥ 0.85%. Specifically, it seems that the presence of PD-1+ TAMs is significantly higher in patients with lymph node metastasis and at age 75 or more. The presence of PD-1+ TAMs in patients aged 75 or more is 3.31%, but the presence of PD-1+ TAMs in patients aged lower than 75 is 1.42%. Furthermore, the percentage of TAMs in patients with lymph node metastasis and in those without lymph node metastasis is 3.83% and 0.54%, respectively [46][32]. Available studies indicate that the prognosis of GC patients is correlated with the expression of the PD-1+ TAM axis. In GC, the expression of PD-1+ TAMs seems to be associated with disease progression and early recurrence in those patients. In Kono et al.’s study, the five-year disease-specific survival rates among patients with GC cells expressing high PD-1+ TAMs and low PD-1+ TAMs are 65.8% and 85.9%, respectively. The comparison of the phenotypic features of GC-infiltrating PD-1+ TAMs revealed that PD-1+ TAMs are polarized to the M2-like type, expressing higher levels of M2 markers such as IL-10 and CCL1. It was also shown that the phagocytotic ability of PD-1+ TAMS is lower than PD-1- TAMs [44][26]. Overall, it seems that PD-1+ TAMs have a pro-tumorigenic activity because they reduce the proliferation of CD8 T cells. On the other hand, PD-1+ TAMs do not correlate with the activity of CD4 T cells [47,48][33][34]. Furthermore, Wei et al. observed that TAMs within the TME exhibited a high degree of PD-L1 expression. Additionally, more infiltration of macrophages was associated with poor prognosis [48][34]. Moreover, a strong correlation was identified between increased macrophage accumulation at the time of diagnosis and an unfavorable prognosis among patients with GC [46,47,48][32][33][34]. Specifically, a correlation was found between a higher accumulation of macrophages in tumors at the time of diagnosis and a poorer prognosis in GC patients. These results suggest that tumors tend to advance preferentially in the presence of a heightened macrophage infiltrate. Conversely, tumors exhibiting a favorable T-cell immunophenotype may experience partial restraint from the immune system, resulting in less frequent progression to advanced stages [49][35].References
- International Agency for Research on Cancer; World Health Organization. The Global Cancer Observatory. Stomach 2020. Available online: https://www.wcrf.org/ (accessed on 10 December 2023).
- González, C.A.; Agudo, A. Chapter 1-Epidemiology, Risk Factors and Pathogenesis, ESMO Gastrointestinal Tract Tumours 2021. Available online: https://oncologypro.esmo.org (accessed on 12 December 2023).
- Rihawi, K.; Ricci, A.D.; Rizzo, A.; Brocchi, S.; Marasco, G.; Pastore, L.V.; Lorena, F.; Llimpe, R.; Golfieri, R. Tumor-Associated Macrophages and Inflammatory Microenvironment in Gastric Cancer: Novel Translational Implications. Int. J. Mol. Sci. 2021, 22, 3805.
- Taieb, J.; Bennouna, J.; Llorca, F.P.; Basile, D.; Samalin, E.; Zaanan, A. Treatment of gastric adenocarcinoma: A rapidly evolving landscape. Eur. J. Cancer 2023, 195, 113370.
- Guan, W.L.; He, Y.; Xu, R.H. Gastric cancer treatment: Recent progress and future perspectives. J. Hematol. Oncol. 2023, 16, 57.
- Depotte, L.; Palle, J.; Rasola, C.; Broudin, C.; Afrăsânie, V.A.; Mariani, A.; Zaanan, A. New developments and standard of care in the management of advanced gastric cancer. Clin. Res. Hepatol. Gastroenterol. 2023, 11, 102245.
- Zhao, Y.; Bai, Y.; Shen, M.; Li, Y. Therapeutic strategies for gastric cancer targeting immune cells: Future directions. Front. Immunol. 2022, 13, 992762.
- Hashimoto, I.; Oshima, T. Claudins and Gastric Cancer: An Overview. Cancers 2022, 14, 290.
- Li, J. Targeting claudins in cancer: Diagnosis, prognosis and therapy. Am. J. Cancer Res. 2021, 11, 3406–3424.
- Giulia Grizzi, G.; Venetis, K.; Denaro, N.; Bonomi, M.; Celotti, A.; Pagkali, A.; Hahne, J.C.; Tomasello, G.; Petrelli, F.; Fusco, N.; et al. Anti-Claudin Treatments in Gastroesophageal Adenocarcinoma Mainstream and Upcoming Strategies. J. Clin. Med. 2023, 12, 2973.
- Shah, M.A.; Shitara, K.; Ajani, J.A.; Bang, Y.J.; Enzinger, P.; Ilson, D.; Lordick, F.; Van Cutsem, E.; Plazas, J.G.; Huang, J.; et al. Zolbetuximab plus CAPOX in CLDN18.2-positive gastric or gastroesophageal junction adenocarcinoma: The randomized, phase 3 GLOW trial. Nat. Med. 2023, 29, 2133–2141.
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Bragagnoli, A.C.; et al. Nivolumab plus chemotherapy versus chemotherapy as first line treatment for advanced gastric cancer/gastroesophageal junction cancer/esophageal adenocarcinoma (CheckMate 649): A multicenter, randomized, open-label, phase 3 trial. Lancet 2021, 398, 27–40.
- Bang, Y.J.; Kang, Y.K.; Catenacci, D.V.; Muro, K.; Fuchs, C.F.; Geva, R.; Hara, H.; Golan, T.; Garrido, M.; Jalal, S.I.; et al. Pembrolizumab alone or in combination with chemotherapy as first line therapy for patients with advanced gastric or gastroesophageal junction adenocarcinoma: Results from the phase II nonrandomized KEYNOTE-059 study. Gastric Cancer 2019, 22, 828–837.
- Janjigian, Y.Y.; Kawazoe, A.; Bai, Y.; Xu, J.; Lonardi, S.; Metges, J.P.; Yanez, P.; Wyrwicz, L.S.; Shen, L.; Ostapenko, Y.; et al. Pembrolizumab plus trastuzumab and chemotherapy for HER2-positive gastric or gastro-oesophageal junction adenocarcinoma: Interim analyses from the phase 3 KEYNOTE-811 randomised placebo-controlled trial. Lancet 2023, 402, 10418.
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489.
- Mazzarella, L.; Duso, B.A.; Trapani, D.; Belli, C.; D’Amico, P.; Ferraro, E.; Viale, G.; Curigliano, G. The evolving landscape of’ next-generation’ immune checkpoint inhibitors: A review. Eur. J. Cancer 2019, 117, 14–31.
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, S.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526.
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in cancer immunotherapy: Clinical implications and future considerations. Hum. Vaccin. Immunother. 2019, 15, 1111–1122.
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.X.; Weissman, I.L. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 568–586.
- Takei, S.; Kawazoe, A.; Shitara, K. The New Era of Immunotherapy in Gastric Cancer. Cancers 2022, 14, 1054.
- Ubukata, Y.; Ogata, K.; Sohda, M.; Yokobori, T.; Shimoda, Y.; Handa, T.; Nakazawa, N.; Kimura, A.; Kogure, N.; Sano, A.; et al. Role of PD-L1 Expression during the Progression of Submucosal. Gastric Cancer. Oncol. 2021, 99, 15–22.
- Ju, X.; Zhang, H.; Zhou, Z.; Chen, M.; Wang, Q. Tumor-associated macrophages induce PD-L1 expression in gastric cancer cells through IL-6 and TNF-α signaling. Exp. Cell Res. 2020, 396, 112315.
- Junttila, A.; Helminen, O.; Väyrynen, J.P.; Ahtiainen, M.; Kenessey, I.; Jalkanen, S.; Mecklin, J.P.; Kellokumpu, I.; Kuopio, T.; Böhm, J.; et al. Immunophenotype based on inflammatory cells, PD-1/PD-L1 signaling pathway and M2 macrophages predicts survival in gastric cancer. Br. J. Cancer 2020, 123, 1625–1632.
- Huang, Y.K.; Wang, M.; Sun, Y.; Di Costanzo, N.; Mitchell, C.; Achuthan, A.; Hamilton, J.A.; Busuttil, R.A.; Boussioutas, A. Macrophage spatial heterogeneity in gastric cancer defined by multiplex immunohistochemistry. Nat. Commun. 2019, 10, 3928.
- Harada, K.; Dong, X.; Estrella, J.S.; Correa, A.M.; Xu, Y.; Hofstetter, W.L.; Sudo, K.; Onodera, H.; Suzuki, K.; Suzuki, A.; et al. Tumor-associated macrophage infiltration is highly associated with PD-L1 expression in gastric adenocarcinoma. Gastric Cancer 2018, 21, 31–40.
- Zhang, H.; Li, R.; Cao, Y.; Gu, Y.; Lin, C.; Liu, X.; Lv, K.; He, X.; Fang, H.; Jin, K.; et al. Poor Clinical Outcomes and Immunoevasive Contexture in Intratumoral IL-10-Producing Macrophages Enriched Gastric Cancer Patients. Ann. Surg. 2022, 275, e626–e635.
- Ivanović, T.; Božić, D.; Benzon, B.; Čapkun, V.; Vukojević, K.; Durdov, G.M. Histological Type, Cytotoxic T Cells and Macrophages in the Tumor Microenvironment Affect the PD-L1 Status of Gastric Cancer. Biomedicines 2023, 11, 709.
- Saqib, U.; Sarkar, S.; Suk, K.; Mohammad, O.; Baig, M.S.; Savai, R. Phytochemicals as modulators of M1-M2 macrophages in inflammation. Oncotarget 2018, 9, 17937–17950.
- Narita, Y.; Muro, K. Updated Immunotherapy for Gastric Cancer. J. Clin. Med. 2023, 12, 2636.
- Pan, Y.; Yinda, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084.
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Zin, A.A.M.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2020, 9, 1512.
- Kono, Y.; Saito, H.; Miyauchi, W.; Shimizu, S.; Murakami, Y.; Shishido, Y.; Miyatani, K.; Matsunaga, T.; Fukumoto, Y.; Nakayama, Y.; et al. Increased PD-1-positive macrophages in the tissue of gastric cancer are closely associated with poor prognosis in gastric cancer patients. BMC Cancer 2020, 20, 175.
- Wang, F.; Li, B.; Wei, Y.; Zhao, Y.; Wang, L.; Zhang, P.; Yang, J.; He, W.; Chen, H.; Jiao, Z.; et al. Tumor-derived exosomes induce PD1+ macrophage population in human gastric cancer that promotes disease progression. Oncogenesis 2018, 7, 41.
- Wei, Y.; Zhang, J.; Fan, X.; Zheng, Z.; Jiang, X.; Chen, D.; Lu, Y.; Li, Y.; Wang, M.; Hu, M.; et al. Immune Profiling in Gastric Cancer Reveals the Dynamic Landscape of Immune Signature Underlying Tumor Progression. Front. Immunol. 2022, 13, 935552.
- Huck, B.R.; Kötzner, L.; Urbahns, K. Small Molecules Drive Big Improvements in ImmunoOncology Therapies. Angew. Chem. Int. Ed. Engl. 2018, 57, 4412–4428.
More