Erythropoietic protoporphyria (EPP) is a genetic disorder stemming from reduced ferrochelatase expression, the final enzyme in the pathway of heme biosynthesis. A closely related condition, X-linked protoporphyria (XLP), bears similar clinical features although it arises from the heightened activity of δ-aminolevulinic acid synthase 2 (ALAS2), the first and normally rate-controlling enzyme in heme biosynthesis in developing red blood cells. Both of these abnormalities result in the buildup of protoporphyrin IX, leading to excruciating light sensitivity and, in a minority of cases, potentially fatal liver complications. Traditionally, managing EPP and XLP involved sun avoidance. However, the emergence of innovative therapies, such as dersimelagon, is reshaping the therapeutic landscape for these conditions.
- erythropoietic protoporphyria
- X-linked protoporphyria
- oral
- dersimelagon
1. Introduction
2. Previous Therapies
Behavioral modification is typically advised as first-line treatment, such as sun avoidance, use of zinc oxide sunblock, and protective clothing. However, this is insufficient due to the infeasibility of sun avoidance and indoor exposure to damaging wavelengths of sunlight passing through windows or arising from artificial light sources. Additionally, most sunscreens are more protective against lower wavelength UV radiation and not against the frequencies that cause EPP symptoms [18][19]. Narrowband ultraviolet B phototherapy has been attempted as a prophylactic measure in EPP. There were proposals for patients with EPP/XLP to purposely experience controlled and gradually increasing UVB radiation to produce skin thickening with increased melanin production. This controlled form of UVB radiation requires administration by a trained professional and may be inconvenient for patients, as it involves exposure at least three times weekly for at least five weeks. A small European study with 12 patients noted that it was ineffective in five of these patients, not meeting any statistical or clinical significance [19][20]. Recent EPP Management Guidelines still advise patients to wear opaque clothing and utilize light filters when feasible to minimize phototoxic symptoms [17]. Subsequently, there has been a treatment goal to induce eumelanogenesis without exposure to UV radiation. Early studies of beta-carotene as a treatment option necessitated daily high doses (a minimum of 180 mg/day for adults) for at least three months to reach carotenoid blood levels of at least 800 μg/dL [20][21][22][21,22,23]. Evaluation of these studies found data suggesting that the beta-carotene treatment efficacy was contradictory and that efficacy inversely correlated with study quality [23][24]; that is, a study was unable to find a significant difference between beta-carotene and placebo for photosensitivity in patients with EPP [24][25]. As reviewed in recent guidelines, previous therapies including, although not limited to, cimetidine, isoniazid, or pyridoxine, were not found to have clear benefits [17]. Additionally, there has not been clinical success with the use of vitamin C or N-acetyl cysteine for the treatment of EPP [25][26][27][26,27,28]. Treatment with cimetidine has been purported to inhibit delta-aminolevulinic acid [ALA] synthase; its use has therefore been proposed to reduce PPIX levels in patients with EPP. Though data are limited to case reports or small series without placebo controls or double-blind designs, there are some suggestions that photosensitivity may be decreased with cimetidine treatment [2]. An ongoing phase 2, double-blind, placebo-controlled cross-over trial of cimetidine is currently underway in the United States (NCT05020184). Bitopertin is another oral agent undergoing a phase 2 clinical trial for the treatment of EPP (NCT05308472). As an oral selective glycine transport inhibitor, this agent restricts glycine uptake into erythroid cells. Glycine and succinyl-CoA are the substrates for ALA synthase, the first and rate-controlling enzyme of the PPIX and heme biosynthetic pathway. Afamelanotide (Scenesse, Clinuvel Pharma) was found to provide benefits in EPP and XLP due to its ability to increase eumelanin production by melanocytes, with an expected reduction in the severity of phototoxic reactions to sunlight or other strong light. Afamelanotide has little effect, if any, on the ongoing overproduction of PPIX [28][29]. It was approved in Europe by the European Medicines Agency and in the US by the FDA in 2019 as the first effective medical treatment for EPP [29][30][30,31]. Afamelanotide, [Nle4, D-Phe7]-a-MSH], is an analog of a-MSH, which stimulates the production of eumelanin as a non-selective agonist of melanocortin receptors. It has higher potency and stability than the natural MSH peptide. A recent observational animal study from Switzerland suggests the occurrence of a dose-dependent protective effect from liver damage related to EPP [31][32]. Afamelanotide is administered as a subcutaneous implant, which is injected every two months and gradually releases the active peptide. This treatment was found to reduce severe phototoxic reactions and improve quality of life; however, a trained professional is needed to administer the treatment. Nausea and loss of appetite are the most notable side effects, which may be due to the molecule’s ability to activate other MCRs non-selectively [32][33][34][33,34,35]. In May 2023, a study outlined challenges with obtaining afamelanotide for EPP or XLP [35][36]; notably, there are concerns regarding patient accessibility (Clinuvel Pharma is currently supplying afamelanotide to only a few selected centers) and costs (currently prices in the US are ~$55,000/implant) [35][36]. Thus, its use has been restricted to relatively few patients with EPP/XLP. Dersimelagon (Figure 1) is an orally active small-molecule selective melanocortin-1 receptor agonist that increases the production of eumelanin, resulting in increased skin pigmentation and anti-inflammatory effects, resulting in increased duration of symptom-free sunlight exposure.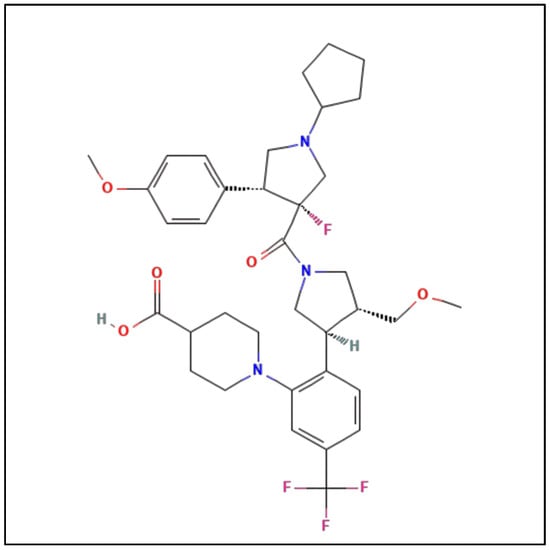
3. Metabolism and Pharmacokinetics of Dersimelagon
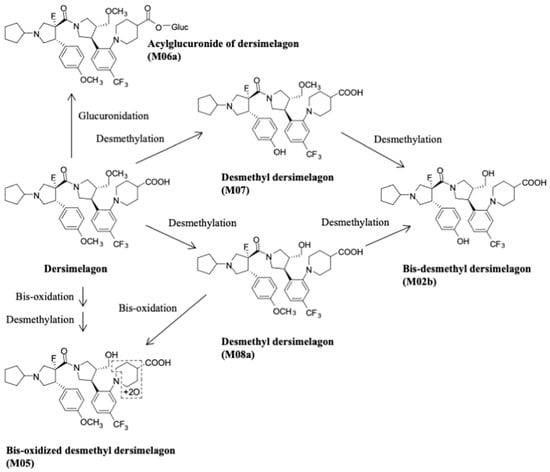
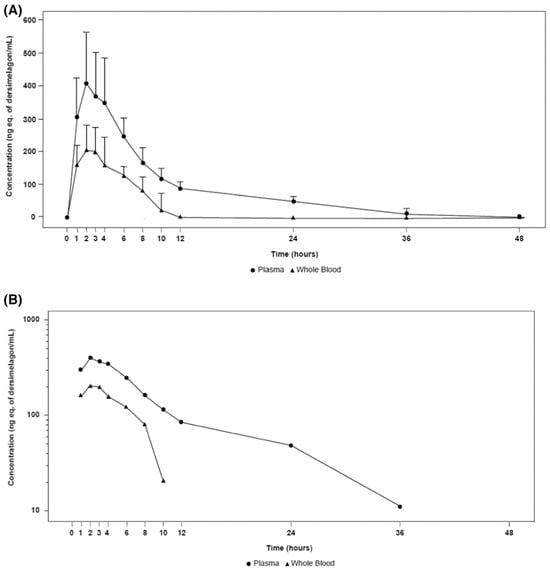
A key component of the appeal of dersimelagon is its oral bioavailability. Mitsubishi Tanabe Pharma Corporation has developed modification methods to improve its stability as a non-peptide [37]. Its molecular structure (Figure 1 [36][18]) has an extensive terminal modification to strengthen and protect the molecule from rapid degradation before reaching its target receptors. In contrast, afamelanotide is unsuitable for oral administration due to the liability of its breakdown in the small intestine by proteases and peptidases, as well as its large size, impairing absorption from the GI tract [37][38][37,38]. The molecular stability of dersimelagon was examined, with an assessment of the oral bioavailability and pharmacokinetics of dersimelagon tablets under a variety of gastric conditions such as a fed state, fasting state, acidic beverage consumption, using high-fat meals, and with a proton pump inhibitor (PPI) [39]. The 50-participant study was an open-label, multicenter, randomized, two-cohort, sequential, and cross-over study in which participants were randomized into two cohorts to receive 300 mg or 100 mg tablets under three gastric conditions: fasted (10+ h), fed (high-fat breakfast with and without esomeprazole 40 mg 2 h before breakfast) with water or a caffeine-free acidic carbonated beverage (355 mL of Canada Dry© ginger ale at a pH of 2.8). No effect was observed on the overall exposure following consumption of a high-fat meal, and Cmax was higher (22%, 90% confidence interval (CI) 1.05–1.42) in a fed state compared with fasted conditions. Similarly, the overall exposure AUC of dersimelagon was comparable following administration alone or in combination with esomeprazole; however, coadministration of esomeprazole led to a slight decrease in Cmax (fasted: 9%, 90% CI 0.77–1.07; fed: 24%, 90% CI 0.66–0.88) compared with the administration of dersimelagon alone. In general, the consumption of an acidic beverage increased the time to Cmax regardless of fed or fasted status and decreased the overall exposure to AUC and Cmax of dersimelagon [39]. The absorption, metabolism, and excretion of dersimelagon in rats, monkeys, and six humans (white males aged 30–65 years, weights 60–110 kg, and with BMI of 18–32 kg/m2, who were overall healthy and free from illness or disease) has been evaluated [36][18]. Participants received a single oral 100 mg dose of radioactive [14C] dersimelagon. Blood, urine, and fecal samples were collected periodically to evaluate Cmax, Tmax, AUC, apparent t1/2, and terminal elimination rate constant (Kel). The median Tmax was two hours in humans [36][18]. As shown in Figure 2 [36][18], dersimelagon is extensively metabolized to glucuronide in the liver, which is eliminated in the bile. In the intestine, some of the glucuronide is metabolized back to the parent drug. In humans, elimination of radioactivity in urine was negligible (excretion of radioactivity into the urine: 0.31% of dose), and the primary route of excretion was feces, with more than 90% of the radioactivity recovered through five days post-dose. Based on these findings, it was concluded that a significant amount of radioactivity from [14C] dersimelagon was not retained in the human body, as shown below (Figure 3 adopted from Suzuki with the axes font increased for improved visibility while maintaining the integrity of the primary literature) [36][18].