Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Sebastian Hasselbeck.
In the rapidly evolving landscape of genetic engineering, the advent of CRISPR-Cas technologies has catalyzed a paradigm shift, empowering scientists to manipulate the genetic code with unprecedented accuracy and efficiency. Despite the remarkable capabilities inherent to CRISPR-Cas systems, recent advancements have witnessed the integration of small molecules to augment their functionality, introducing new dimensions to the precision and versatility of gene editing applications.
- Cas9
- small molecules
- genome editing
1. Introduction
In bacteria and archaea, an important part of their immune systems are the clustered regulatory interspaced short palindromic repeats (CRISPRs) [1]. These are utilized to protect the host organism from invading viruses and plasmids. Within this system, the nucleic acids of intruders are silenced by specific small ribonucleic acids (RNAs) originating from the host organism itself [2]. Over recent years, scientific advancements have transformed this system into a practical tool for (epi)genome editing, organismal studies, and the exploration and combat of diseases, particularly hereditary diseases [3]. Playing a pivotal role in the CRISPR system are the CRISPR-associated proteins (Cas) [4]. Among them, Cas9 is the most widely used [5]. It functions as an endonuclease, capable of recognizing specific double-stranded DNA (dsDNA) and, in turn, silencing it through cleavage [2]. Moreover, in scientific applications, after dsDNA cleavage, a new sequence can be inserted to achieve designated genome editing [3]. However, low editing efficiency and unwanted off-target effects largely limit clinical applications of the CRISPR/Cas9 system [6].
2. Small Molecules Modulate Wild-Type Cas9 Protein
Through high-throughput screening, Maji et al., identified several small molecules that could disrupt Cas9 binding to DNA, preventing DNA double-strand breaks (DSBs). Testing eGFP further confirmed that some of these Cas9 inhibitors, such as BRD0539 (1), worked reversibly [37,38][7][8]. Recently, using a high-content fluorescence-based approach, the researchers identified valproic acid (2) (VPA) as a Cas9 degrader from a chemical library consisting of nearly 300 drug-like compounds and natural products [39][9]. VPA, a well-known histone deacetylase inhibitor (HDACi), demonstrated significant Cas9WT degradation under hyperthermia conditions generated either in the presence of a photothermal agent, indocyanine green, upon irradiation by a near-infrared laser or heating with an external heat bag. This degradation effect was independent of its HDAC inhibitory effect. However, the off-target effects of BRD0539 or VPA remain unknown. For a clearer understanding of off-target effects, Yang et al. searched for small molecules that inhibit Cas9WT. In their studies, the most effective Cas9 inhibitor was shown to be SP24 (3), with an IC50 for Cas9WT of approximately 14 µM and for the Cas9-sgRNA complex of about 7 µM. This was shown during an FP assay, where SP24 significantly decreased fluorescence polarization [38][8]. While there are very much more potent inhibitors (nM range) [40][10], the SP inhibitors showed higher IC50 values for Cas9wt than the previously known BRD0539 inhibitor with an IC50 of 22 µM [37][7]. And even though the IC50 may not be optimal, the advantage is that the inhibitor can be applied to Cas9wt. The upcoming discussions will emphasize the often-severe modifications of Cas9 to make it addressable by small molecules. Furthermore, SP24 and SP2 (4) were proven to enhance the precision of Cas9-mediated genome editing (Figure 61) [37,38][7][8].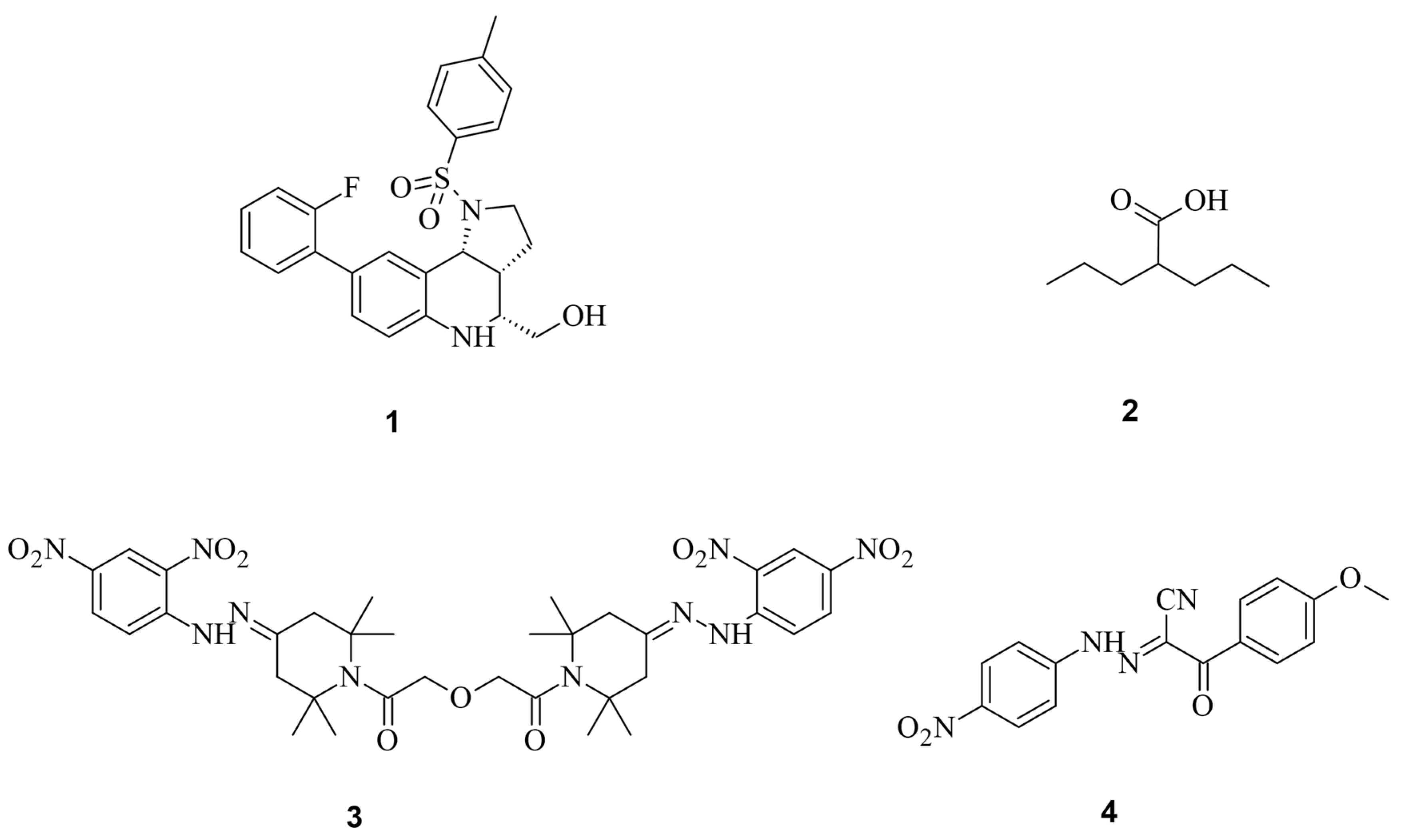
Figure 61.
The spCas9 small molecules inhibitors and degraders BRD0539 (
1
), VPA (
2
), SP24 (
3
), and SP2 (
3. Small Molecules Modulate Engineered Cas9 Protein
While only a few small molecules have been identified to function with Cas9WT, the primary Cas9 chemical modulators interact with engineered Cas9. A way to activate Cas9 with a small molecule was demonstrated by Davis et al. [41][11]. As shown in Figure 72, Buskirk et al. successfully evolved inteins that could only self-remove in the presence of 4-hydroxytamoxifen (4-HT) (5) [42][12].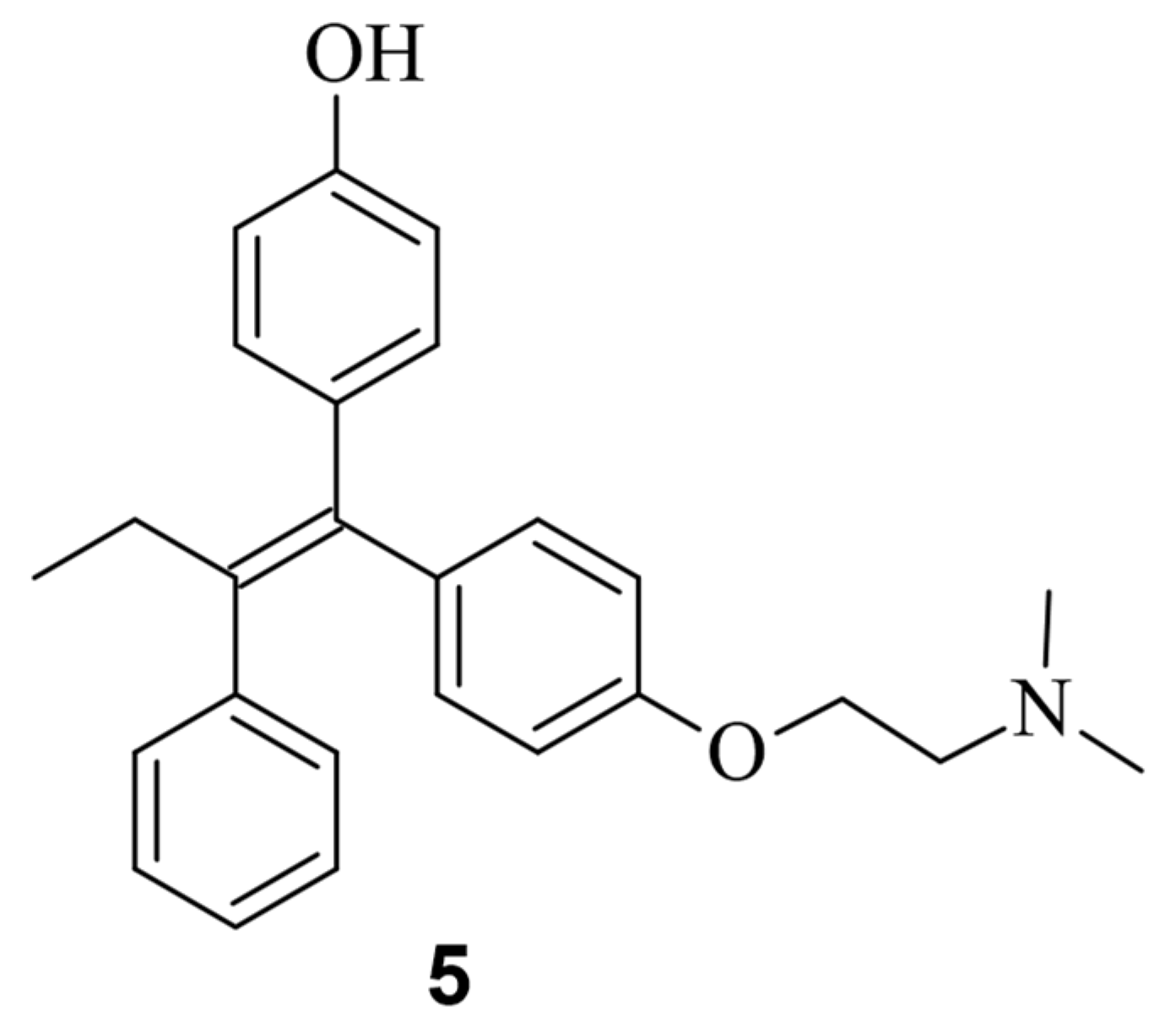
Figure 72.
Chemical structure of the small molecule 4-hydroxytamofen (
5
).
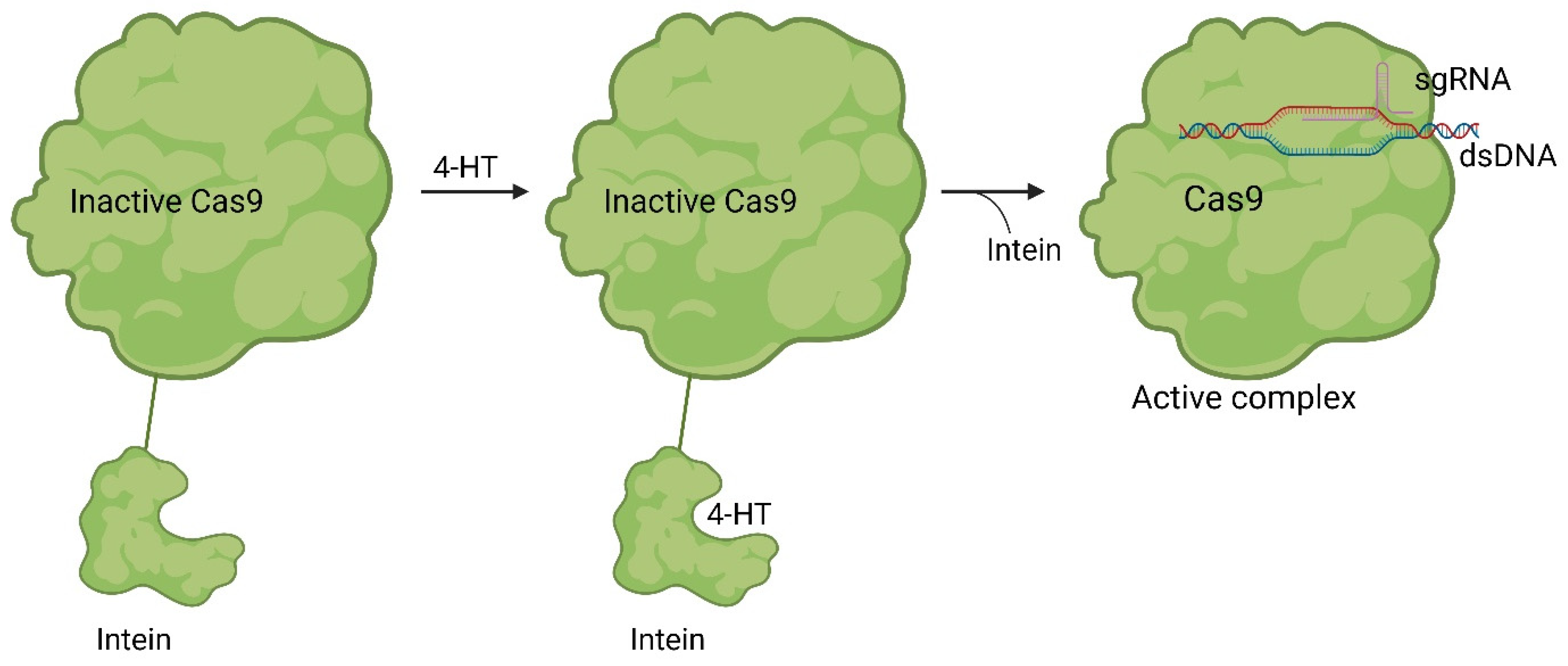
Figure 83. Working mode of intein inhibited and activated Cas9. Through a modification with an intein, Cas9 is inactivated (left). Through treatment with the small molecule 4-HT, self-splicing of the intein is induced (middle). After splicing, Cas9 can form an active sgRNA complex, and the desired gene can be modified [41][11].
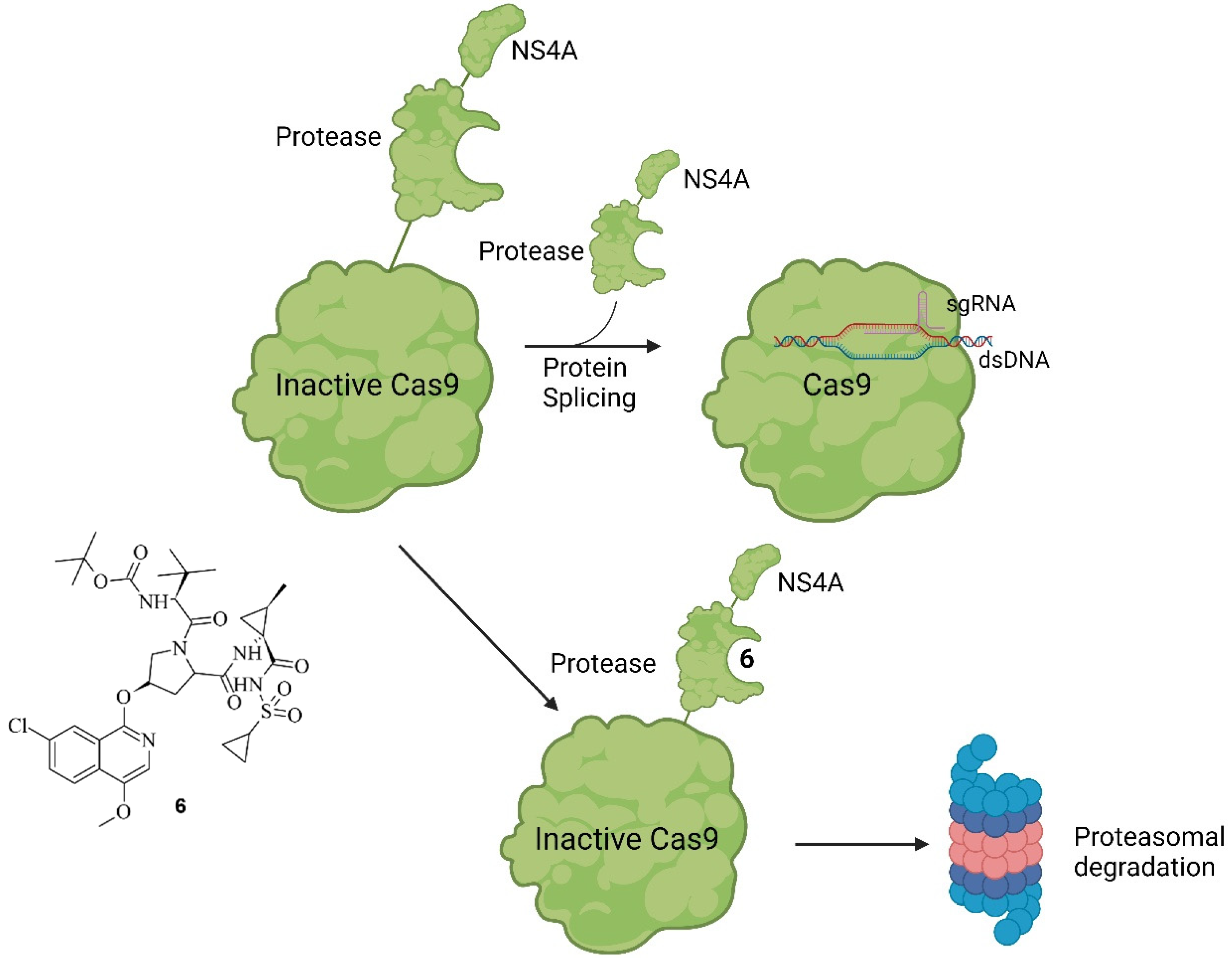
Figure 94. Working principle of SMASh tag-controlled Cas9. Under non-treatment conditions, modified Cas9 will be processed into active Cas9 by self-cleavage of the SMASh tag. The tag will be degraded by the proteasome or lysosome. If, however, treatment with ASV (6) (shown on the left) is applied, the protease activity is inhibited, and Cas9 marked with a SMASh tag is degraded as a whole unit [43][13].
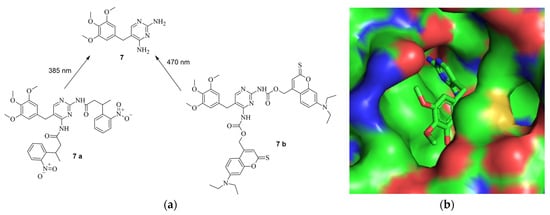
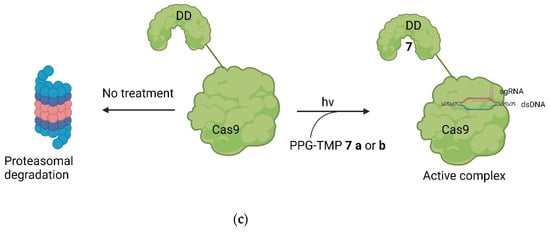
Figure 105. (a) (above, left): The structure of TMP (7) and the protected derivates 7 a and 7 b, including the used wave lengths for deprotection [32][18]. (b) (above, right): The structure of TMP in the binding pocket of DHFR. The amines are deep in the binding pocket, leaving them a good target for protection (PDB: 7R6G) [32,48][18][19]. (c) (below): The principle of the system. Through introduction of the DDs, Cas9 is quickly degraded by the proteosome. By treatment with PPG-TMP and irradiation (hv), free TMP can be formed, bind to the DDs, stabilize them, and thus turn Cas9 active [32][18].
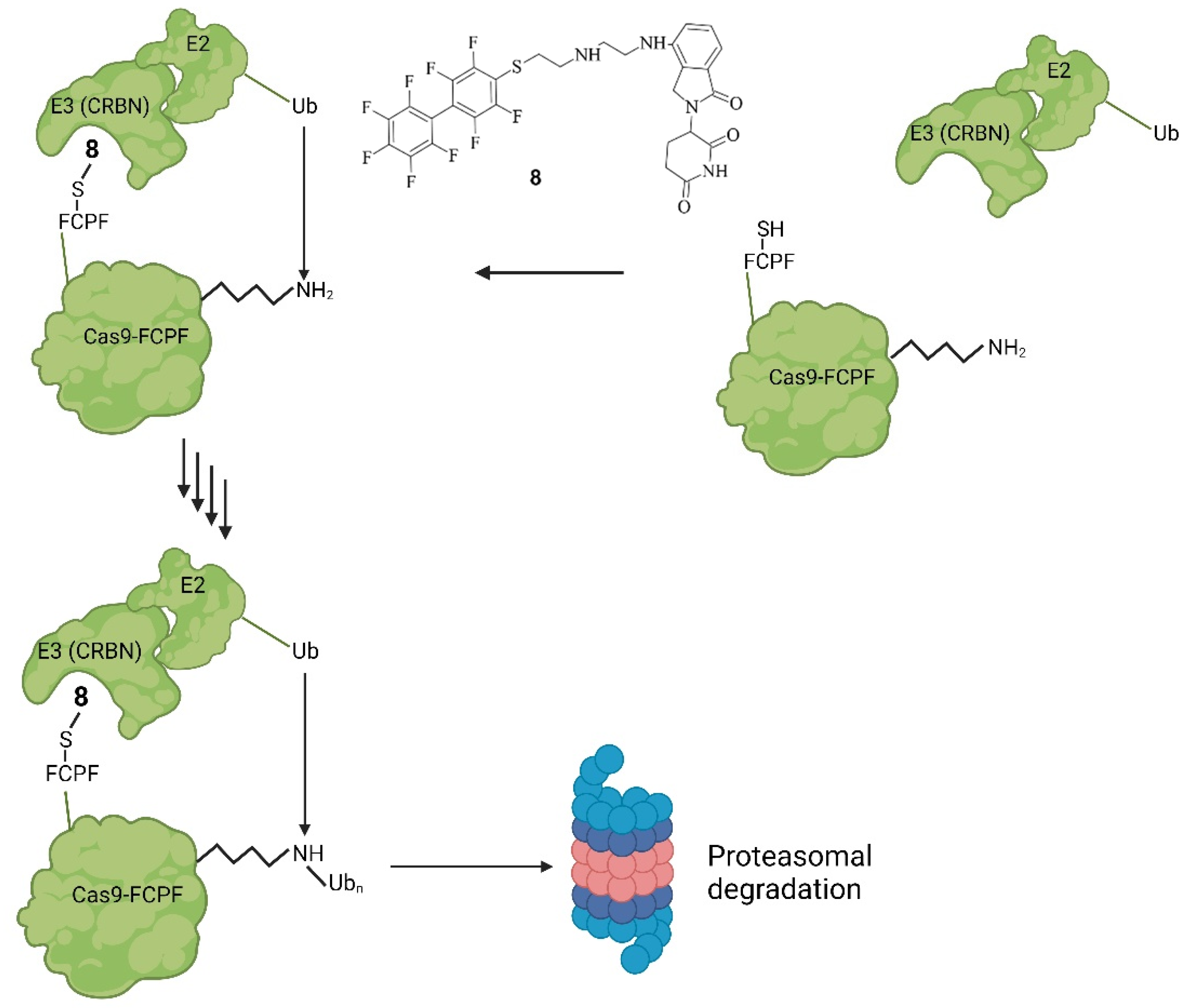
Figure 116. Mode of action of PROTAC-FCPF (8). Binding of 8 onto Cas9-FCPF leads to a complex with the E3-ligase CRBN. Attached to that is an E2-ligase. The ubiquitination of Cas9-FCPF is then catalysed, which ultimately leads to the degradation of Cas9-FCPF by the UPS, rendering Cas9-FCPF inactive [49,51][20][22].
4. Small Molecules Regulate DSB Repair Mechanisms
Aside from regulating the activity of engineered Cas9, there is the possibility of modulating the CRISPR/Cas9 system by regulating the DNA repair system. Li et al. investigated three compounds regarding their ability to enhance HDR or down-regulate NHEJ towards a more precise editing [17][25]. In their study, Scr7 (9), L755507 (10), and Resveratrol (11) were found to inhibit DNA ligase IV and thereby reduce NHEJ-mediated repair [17][25], showing that the efficiency of knock-ins can be enhanced in the presence of chemical inhibitors targeting NHEJ-influencing factors [54][26]. The compounds are illustrated in Figure 127.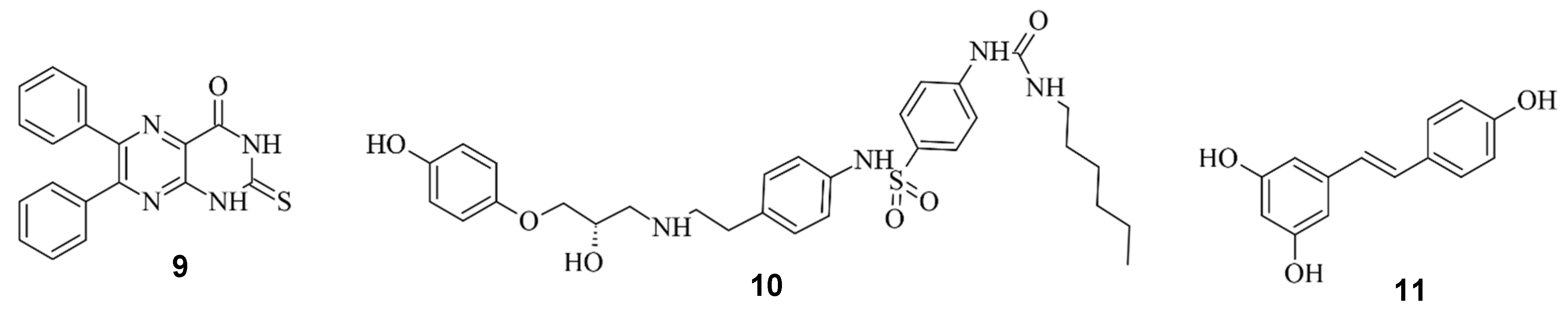
Figure 127.
Scr7 (
9
), L755507 (
10
) and Resveratrol (
11
) from left to right.
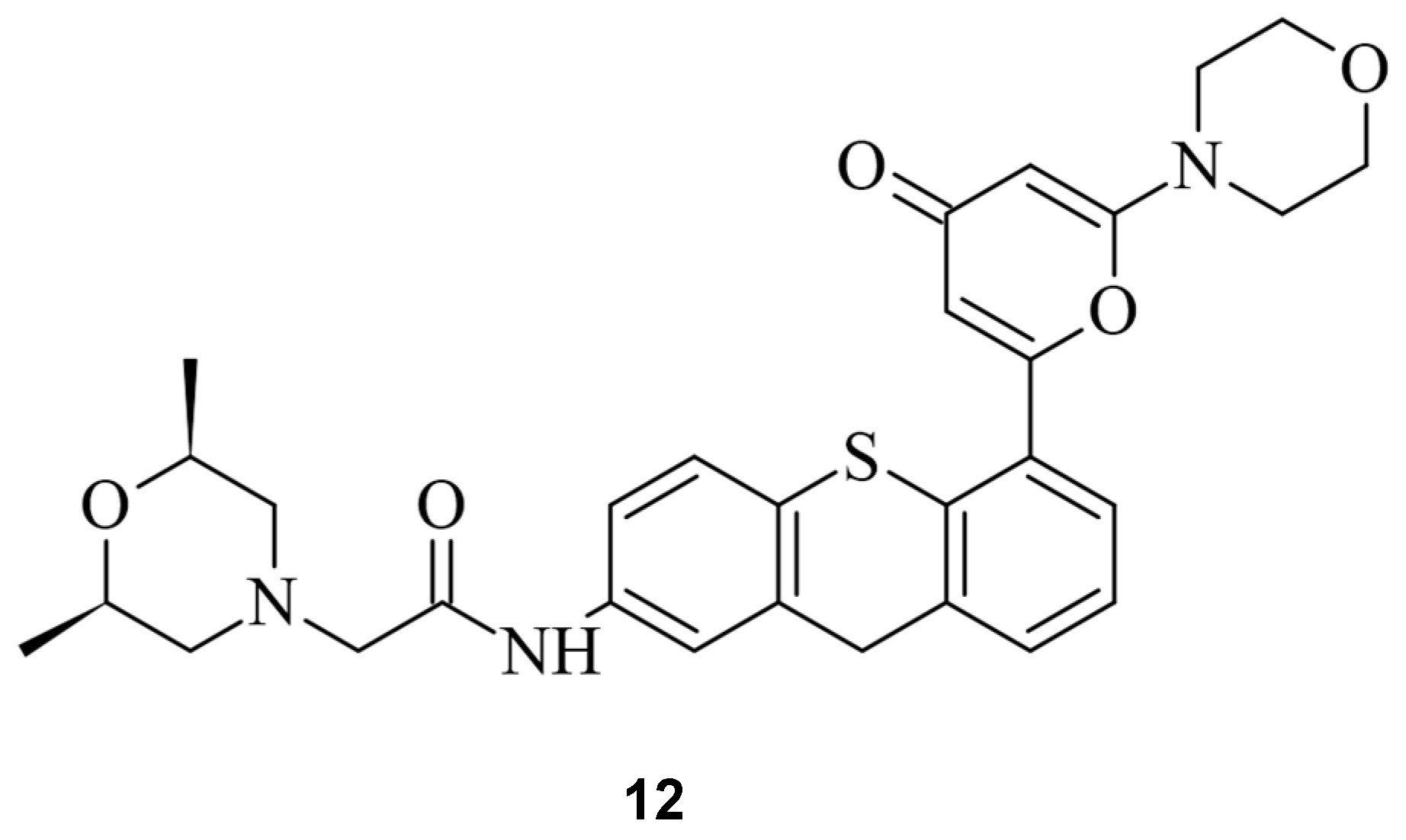
Figure 138.
The ATM inhibitor KU60019 (
12
).
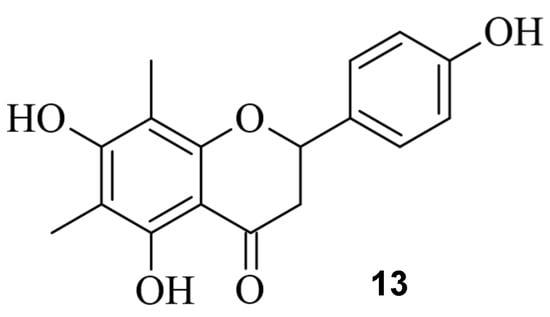
Figure 149.
The small molecule farrerol (
13
).
5. Small Molecules Regulate sgRNAs
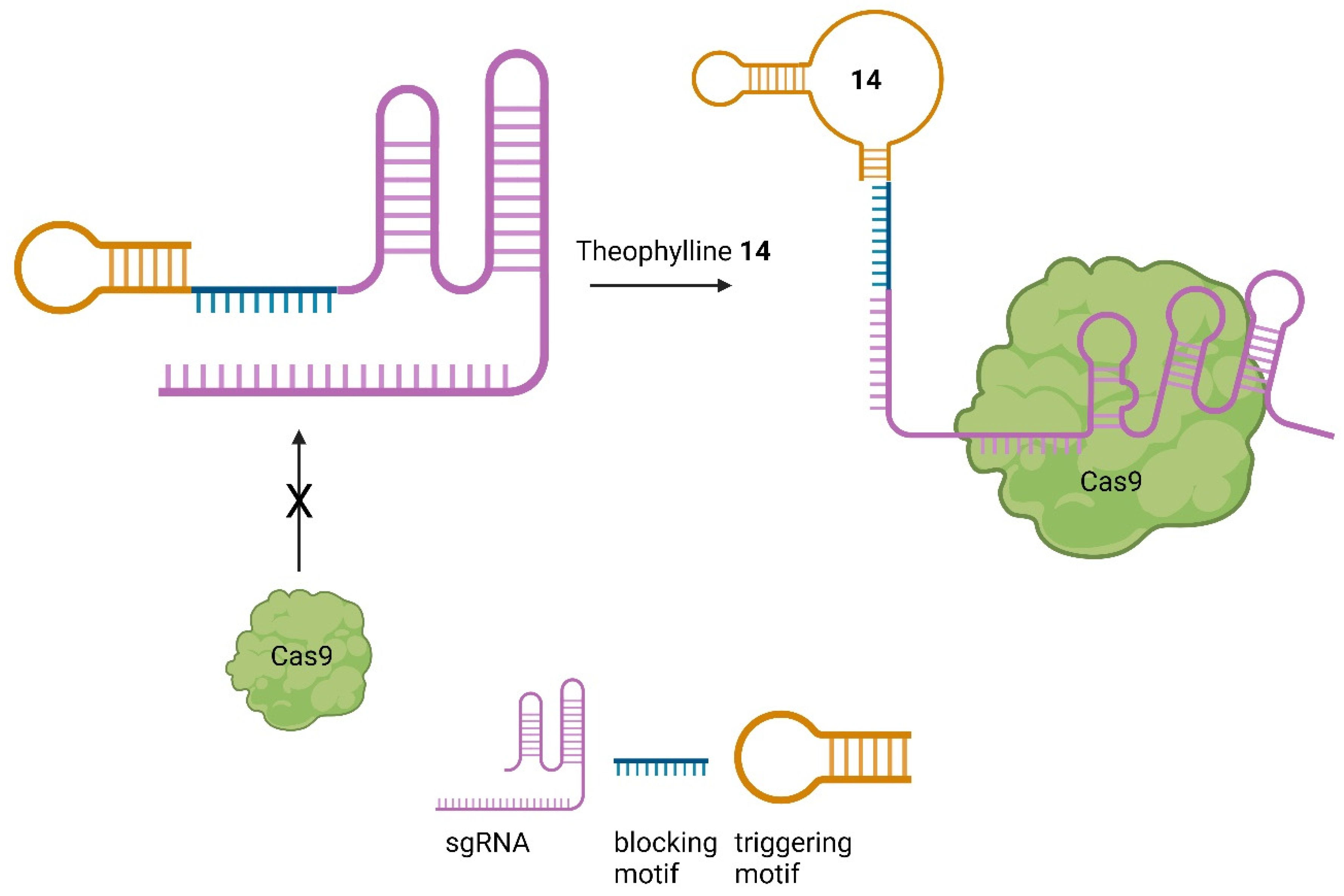
Given the important role of sgRNAs in CRISPR/Cas9 [2], chemicals capable of controlling the activity of the sgRNA offer another possibility to regulate the process of genome editing [41,43][11][13]. Aptamers are short nucleic acids that can specifically bind ligands and are commonly employed to regulate nucleic acids, such as sgRNA [62][34]. Iwasaki et al. applied this concept to develop so-called aptamer-sgRNA (agRNA), in which the theophylline (14) or 3MX (15) aptamer was used and introduced into sgRNA at different positions, leading to an endonuclease-active Cas9 only upon treatment with 14 or 15, respectively. An up to 104-fold increase in transformants in comparison to wild-type sgRNA was observed (Figure 150) [63][35].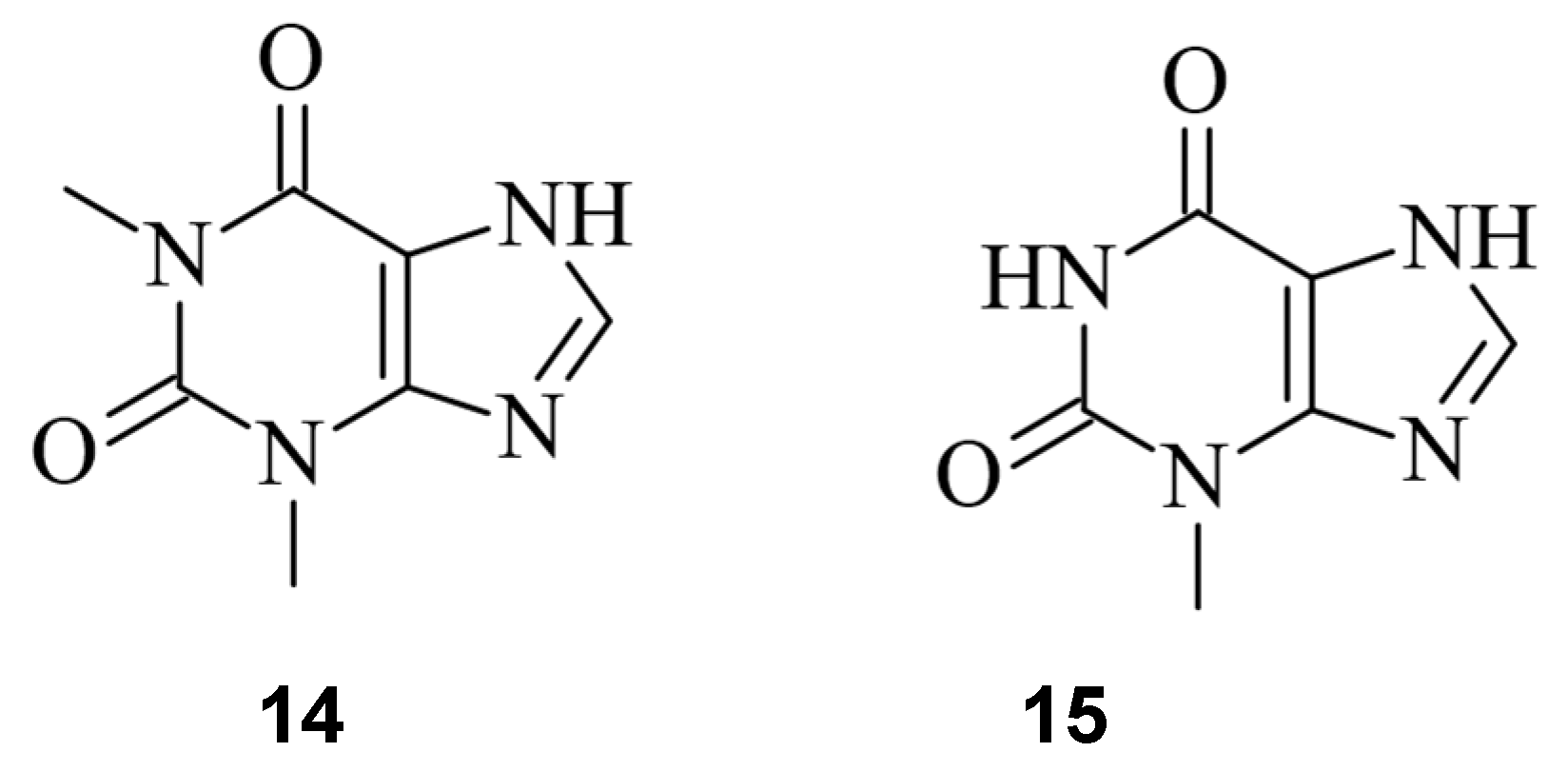
Figure 150.
The RNA aptamer ligands theophylline (
References
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338.
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821.
- Xu, Y.; Li, Z. CRISPR-Cas systems: Overview, innovations and applications in human disease research and gene therapy. Comput. Struct. Biotechnol. J. 2020, 18, 2401–2415.
- Shrock, E.; Güell, M. CRISPR in Animals and Animal Models. Prog. Mol. Biol. Transl. Sci. 2017, 152, 95–114.
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911.
- Lee, S.W.; Tran, K.T.; Vazquez-Uribe, R.; Gotfredsen, C.H.; Clausen, M.H.; Mendez, B.L.; Montoya, G.; Bach, A.; Sommer, M.O.A. Identification and Optimization of Novel Small-Molecule Cas9 Inhibitors by Cell-Based High-Throughput Screening. J. Med. Chem. 2022, 65, 3266–3305.
- Maji, B.; Gangopadhyay, S.A.; Lee, M.; Shi, M.; Wu, P.; Heler, R.; Mok, B.; Lim, D.; Siriwardena, S.U.; Paul, B.; et al. A High-Throughput Platform to Identify Small-Molecule Inhibitors of CRISPR-Cas9. Cell 2019, 177, 1067–1079.e19.
- Yang, Y.; Li, D.; Wan, F.; Chen, B.; Wu, G.; Li, F.; Ren, Y.; Liang, P.; Wan, J.; Songyang, Z. Identification and Analysis of Small Molecule Inhibitors of CRISPR-Cas9 in Human Cells. Cells 2022, 11, 3574.
- Cheng, X. Valproic Acid Thermally Destabilizes and Inhibits SpyCas9 Activity. Mol. Ther. 2020, 28, 2635–2641.
- Crowley, W.R. BIBO3304. Xpharm: Compr. Pharmacol. Ref. 2007, 1, 1–3.
- Davis, K.M.; Pattanayak, V.; Thompson, D.B.; Zuris, J.A.; Liu, D.R. Small molecule–triggered Cas9 protein with improved genome-editing specificity. Nat. Chem. Biol. 2015, 11, 316–318.
- Buskirk, A.R.; Ong, Y.C.; Gartner, Z.J.; Liu, D.R. Directed evolution of ligand dependence: Small-molecule-activated protein splicing. Proc. Natl. Acad. Sci. USA 2004, 101, 10505–10510.
- Wu, Y.; Yang, L.; Chang, T.; Kandeel, F.; Yee, J.K. A Small Molecule Controlled Cas9 Repressible Sysytem. Mol. Ther. Nucleic Acids 2020, 19, 922–932.
- Rose, J.C.; Popp, N.A.; Richardson, C.D.; Stephany, J.J.; Mathieu, J.; Wei, C.T.; Corn, J.E.; Maly, D.J.; Fowler, D.M. Suppression of unwanted CRISPR-Cas9 editing by co-administration of catalytically inactivating truncated guide RNAs. Nat. Commun. 2020, 11, 2697.
- Sekine, R.; Kawata, T.; Muramoto, T. CRISPR/Cas9 mediated targeting of multiple genes in Dictyostelium. Sci. Rep. 2018, 8, 8471.
- Weinstain, R.; Slanina, T.T.; Kand, D.; Kla, P.K. Visible-to-NIR-Light Activated Release: From Small Molecules to Nanomaterials. Chem. Rev. 2020, 120, 13135–13272.
- Maji, B.; Moore, C.L.; Zetsche, B.; Volz, S.E.; Zhang, F.; Shoulders, M.D.; Choudhary, A. Multidimensional chemical control of CRISPR–Cas9. Nat. Chem. Biol. 2016, 13, 9–11.
- Manna, D.; Maji, B.; Gangopadhyay, S.A.; Cox, K.J.; Zhou, Q.; Law, B.K.; Mazitschek, R.; Choudhary, A. A Singular System with Precise Dosing and Spatiotemporal Control of CRISPR-Cas9. Angew. Chem. Int. Ed. 2019, 58, 6285–6289.
- Krucinska, J.; Lombardo, M.N.; Erlandsen, H.; Estrada, A.; Si, D.; Viswanathan, K.; Wright, D.L. Structure-guided functional studies of plasmid-encoded dihydrofolate reductases reveal a common mechanism of trimethoprim resistance in Gram-negative pathogens. Commun. Biol. 2022, 5, 459.
- Gama-Brambila, R.A.; Chen, J.; Dabiri, Y.; Tascher, G.; Němec, V.; Münch, C.; Song, G.; Knapp, S.; Cheng, X. A Chemical Toolbox for Labeling and Degrading Engineered Cas Proteins. JACS Au 2021, 1, 777–785.
- Zhang, C.; Welborn, M.; Zhu, T.; Yang, N.J.; Santos, M.S.; Van Voorhis, T.; Pentelute, B.L. π-Clamp-mediated cysteine conjugation. Nat. Chem. 2016, 8, 120–128.
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200.
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019.
- Bubeck, F.; Hoffmann, M.D.; Harteveld, Z.; Aschenbrenner, S.; Bietz, A.; Waldhauer, M.C.; Börner, K.; Fakhiri, J.; Schmelas, C.; Dietz, L.; et al. Engineered anti-CRISPR proteins for optogenetic control of CRISPR–Cas9. Nat. Methods 2018, 15, 924–927.
- Li, G.; Zhang, X.; Zhong, C.; Mo, J.; Quan, R.; Yang, J.; Liu, D.; Li, Z.; Yang, H.; Wu, Z. Small molecules enhance CRISPR/ Cas9-mediated homology-directed genome editing in primary cells OPEN. Sci. Rep. 2017, 7, 8943.
- Srivastava, M.; Nambiar, M.; Sharma, S.; Karki, S.S.; Goldsmith, G.; Hegde, M.; Kumar, S.; Pandey, M.; Singh, R.K.; Ray, P.; et al. An Inhibitor of Nonhomologous End-Joining Abrogates Double-Strand Break Repair and Impedes Cancer Progression. Cell 2012, 151, 1474–1487.
- Bermudez-Cabrera, H.C.; Culbertson, S.; Barkal, S.; Holmes, B.; Shen, M.W.; Zhang, S.; Gifford, D.K.; Sherwood, R.I. Small molecule inhibition of ATM kinase increases CRISPR-Cas9 1-bp insertion frequency. Nat. Commun. 2021, 12, 5111.
- Ueno, S.; Sudo, T.; Hirasawa, A. ATM: Functions of ATM Kinase and Its Relevance to Hereditary Tumors. Int. J. Mol. Sci. 2022, 23, 523.
- Lombard, D.B.; Chua, K.F.; Mostoslavsky, R.; Franco, S.; Gostissa, M.; Alt, F.W. DNA Repair, Genome Stability, and Aging. Cell 2005, 120, 497–512.
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair. 2008, 7, 1765–1771.
- Chen, Y.; Zhang, H.; Xu, Z.; Tang, H.; Geng, A.; Cai, B.; Su, T.; Shi, J.; Jiang, C.; Tian, X.; et al. A PARP1-BRG1-SIRT1 axis promotes HR repair by reducing nucleosome density at DNA damage sites. Nucleic Acids Res. 2019, 47, 8563–8580.
- Wassing, I.E.; Graham, E.; Saayman, X.; Rampazzo, L.; Ralf, C.; Bassett, A.; Esashi, F. The RAD51 recombinase protects mitotic chromatin in human cells. Nat. Commun. 2021, 12, 5380.
- Zhang, W.; Chen, Y.; Yang, J.; Zhang, J.; Yu, J.; Wang, M.; Zhao, X.; Wei, K.; Wan, X.; Xu, X.; et al. A high-throughput small molecule screen identifies farrerol as a potentiator of CRISPR/Cas9-mediated genome editing. eLife 2020, 9, e56008.
- Meek, K.N.; Rangel, A.E.; Heemstra, J.M. Enhancing aptamer function and stability via in vitro selection using modified nucleic acids. Methods 2016, 106, 29–36.
- Iwasaki, R.S.; Ozdilek, B.A.; Garst, A.D.; Choudhury, A.; Batey, R.T. Small molecule regulated sgRNAs enable control of genome editing in E. coli by Cas9. Nat. Commun. 2020, 11, 1394.
- Wrist, A.; Sun, W.; Summers, R.M. The Theophylline Aptamer: 25 Years as an Important Tool in Cellular Engineering Research. ACS Synth. Biol. 2020, 9, 682–697.
- Lee, S.W.; Zhao, L.; Pardi, A.; Xia, T. Ultrafast dynamics show that the theophylline and 3-methylxanthine aptamers employ a conformational capture mechanism for binding their ligands. Biochemistry 2010, 49, 2943–2951.
- Lin, B.; An, Y.; Meng, L.; Zhang, H.; Song, J.; Zhu, Z.; Liu, W.; Song, Y.; Yang, C. Control of CRISPR-Cas9 with small molecule-activated allosteric aptamer regulating sgRNAs. Chem. Commun. 2019, 55, 12223.
More