MYC amplification or overexpression is most common in Group 3 medulloblastomas and is positively associated with poor clinical outcomes. Protein arginine methyltransferase 5 (PRMT5) overexpression has been shown to be associated with tumorigenic MYC functions in cancers, particularly in brain cancers such as glioblastoma and medulloblastoma. PRMT5 regulates oncogenes, including MYC, that are often deregulated in medulloblastomas. However, the role of PRMT5-mediated post-translational modification in the stabilization of these oncoproteins remains poorly understood. The potential impact of PRMT5 inhibition on MYC makes it an attractive target in various cancers. PRMT5 inhibitors are a promising class of anti-cancer drugs demonstrating preclinical and preliminary clinical efficacies.
- brain cancer
- medulloblastoma
- MYC
- PRMT5 inhibitors
- SDMA
1. Introduction
2. PRMT5 Structure, Function, and Localization
2.1. Structure
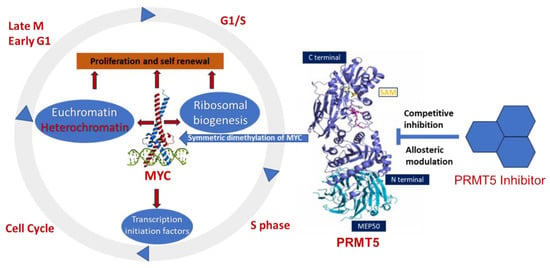
PRMT5 is a primary type II arginine methyl transferase that forms a prominent methylosome complex with distinctive binding oligopeptides, such as the WD (Trp-Asp) repeat-containing 50-kilodalton methylosome protein (MEP50). PRMT5 requires the existence of diverse substrate adapters such as rio-domain-containing protein 1 (RioK1), chloride channel nucleotide-sensitive 1A protein (pIC1n) and cooperator of PRTM5 (COPR5) to detect and catalyze the SDMA on histone and non-histone proteins via PTMs [31][32][33][31,32,33]. PRMT5′s structure consists of a triphosphate isomerase (TIM) barrel, an intermediate Rossmann-fold, and a C-terminal β-barrel [34]. Four PRMT5 units generate a hetero octameric complex by binding with four MEP50s (Figure 1). Studies have demonstrated that PRMT5 alone has minimal methyltransferase activity; it must be complexed with MEP50 to achieve normal catalysis of SDMA on proteins [35]. This could be because MEP50 enhances the stability of PRMT5 for a long time by binding with proteins and acting as a metastable cofactor.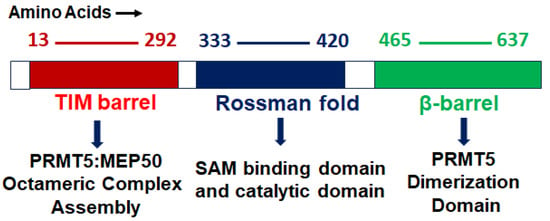
2.2. Function
PRMT5 plays a key role in cell functions and processes by regulating the methylation of cellular proteins, which affects oncogenic cellular processes such as cell proliferation and differentiation [29][30][36][29,30,36]. PRMT5 regulates these processes by modifying gene expression to stabilize histones H4R3, H3R2, H3R8, and H2AR3 and non-histone proteins via the SDMA process [37][38][37,38]. An extensive range of nonhistone proteins have also been revealed as PRMT5 substrates, including androgen receptor (AR), EGFR, GATA4, C-MYC, N-MYC, IL-2, E2F1, GM130, HOXA9, KLF4, KLF5, NOTCH, NFkB(p65), PDCD4, POLR2A, P53, RAF proteins, SPT5, SREBP1a, Sm proteins, nucleolin, and others [12][36][39][40][41][42][43][44][45][46][47][48][49][50][51][52][12,36,39,40,41,42,43,44,45,46,47,48,49,50,51,52]. In addition, some substrates such as certain nonhistone oncogenic transcription factors are symmetrically dimethylated by PRMT5. PRMT5-regulated cellular processes are shown in Figure 2. The importance of arginine methylation by PRMT5 in cancer progression has only recently become apparent [17]. PRMT5 knockout mice exhibited embryonic lethality, demonstrating the role of PRTM5 in embryonic development and crucial biological functions. In mouse embryonic stem cells (ESCs), PRMT5 maintains pluripotency, whereas in human ESCs, it influences only proliferation [53][54][53,54]. PRMT5 is needed for neural stem cell persistence and its deletion causes premature death of the mouse by disrupting the development of the central nervous system [55]. PRMT5 promotes SWI/SNF-mediated chromatin remodeling and controls the process of myogenesis. Deletion of PRMT5 causes an obstacle in developmental processes, uncontrolled proliferation, and impairment of adult tissue differentiation [53][54][55][56][57][58][53,54,55,56,57,58]. Notably, PRMT5 is overexpressed in a number of cancers, including melanoma, multiple myeloma, lymphoma, glioblastoma, breast, lung, pancreas, prostate, ovarian, and colorectal cancers, and high expression of PRMT5 often correlates with poor patient clinical outcomes [38][59][38,59]. Organ-specific functions of PRMT5 are shown in Table 1. The higher expression of PRMT5 in cancer is thought to epigenetically suppress tumor suppressor and cell cycle genes [17][60][17,60]. Recently, the association of PRMT5 with MYC was found in numerous cancers, including brain tumors such as glioblastoma; this association creates abnormalities in MYC function [61][62][63][61,62,63]. Consequently, PRMT5 has been recognized as an oncogenic function and has received extensive interest as a potential target for better clinical outcomes. To this end, numerous potent therapeutic agents have been developed to inhibit PRMT5 and their antitumor effects are now being assessed in preclinical models and clinical trials [27][64][27,64].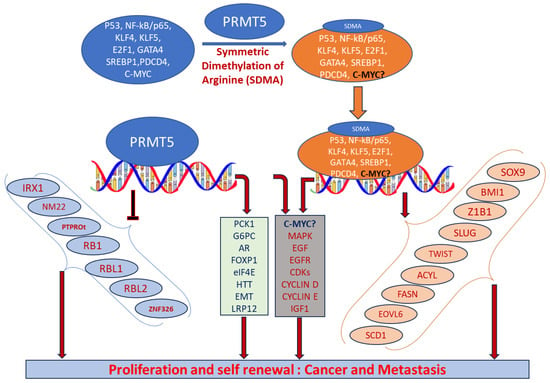
2.3. Localization
Cytosolic and nuclear localization of PRMT5 helps to determine its role in the cell. PRMT5 is predominantly localized in the cytoplasm in lung [65], prostate [66], and melanoma cancer [67]. Diffused cellular localization of PRMT5 was confirmed in both the cytoplasm and the nucleus of brain tumor glioblastoma cells [61]. Cytoplasmic and nuclear localization of PRMT5 has also been confirmed in various preclinical mouse models and primary human cancer tissues [68]. In adult mice, PRMT5 is expressed predominantly in the nucleus of the neurons in the cerebrum and spinal cord [55]. Han et al. demonstrated the high expression of PRMT5 as a marker of malignant progression in glioblastoma and its crucial role in tumor growth [63]. The reseaOurchers' lab recently analyzed the localization of the PRMT5 in tumor tissues of medulloblastoma patients as well as in MYC-amplified cell lines. PRMT5 demonstrated predominantly nuclear localization in both HD-MB03 and primary tumor cells [22].|
Organ |
Cellular Function |
Mechanism |
References |
|---|---|---|---|
|
Brain |
Cell cycle progression, apoptosis |
|
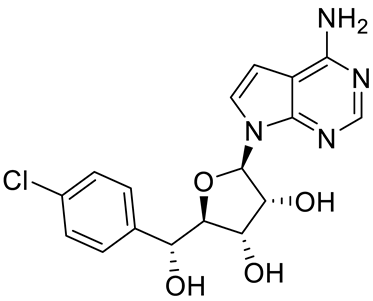
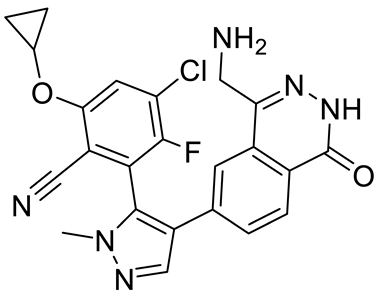
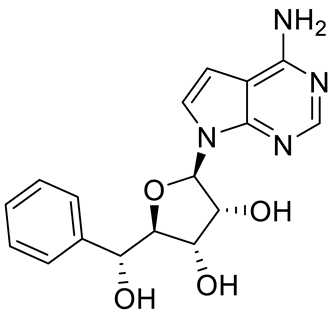
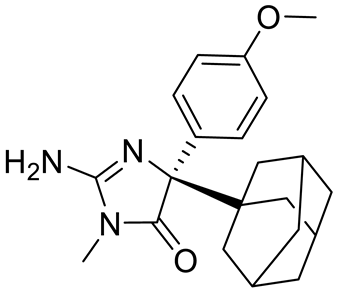
Compound Name |
Structure |
Function |
Status IC50 In Vitro |
In Vivo Activity |
References |
|---|
Abbreviations: AML, acute myeloid leukemia; MCL, mantle cell lymphoma; MM, myelomonocytic leukemia; GBM, glioblastoma; TNBC, triple-negative breast cancer, PDX, patient-derived xenograft; DLBCL, diffuse large B cell lymphoma; CNS, central nervous system; MPN, myeloproliferative neoplasm; nM, nano molar; NA, not available; SAM, S-adenosylmethionine; MTA, methylthioadenosine.
|
ClinicalTrials.gov Identifier |
Name of Inhibitor |
Disease |
|||||||
|---|---|---|---|---|---|---|---|---|---|
|
JNJ64619178 |
|
Altered expression and stability of MYC |
[22] |
||||||
Dual SAM/substrate competitive |
0.2 nM |
||||||||
|
NCT03573310 |
JNJ64619178 | Antitumor effect in lung cancer, AML, non-Hodgkin lymphoma cell line mouse xenograft |
Phase I |
Neoplasm solid tumors, non-Hodgkin lymphoma, and myelodysplastic syndrome [108] |
|||||
|
PF06939999 |
|||||||||
|
NCT03854227 |
Phase separation |
|
PF06939999 Methylation of FUS |
SAM [69] |
|||||
competitive |
Phase I3.3 nM |
Antitumor effect in lung cancer |
[109] |
||||||
Advance and metastatic solid tumors | GSK3β-NF-kβ signaling |
GSK3235025 EPZ015666 |
Altered expression of E2F1 |
Substrate competitive [70] |
|||||
22 nM | Antitumor effect in MCL, MM, AML, GBM, and bladder cell line mouse xenografts and in a TNBC PDX mouse model |
||||||||
|
NCT03614728 |
GSK3326595 |
Phase I and II |
Metastatic solid tumors and acute myeloid leukemia | , |
HTT toxicity |
||||
|
GSK591 (EPZ015866) | Altered expression of HTT |
[71] |
|||||||
|
AKT-ERK signaling, cell cycle progression | |||||||||
|
NCT02783300 |
|
GSK3326595 Substrate competitive |
Phase I |
Altered expression of PTEN |
[72] |
||||
4 nM |
Solid tumors and non-Hodgkin lymphoma |
Cell cycle progression, stemness |
Altered RNA Splicing |
||||||
Antitumor effect in glioblastoma | [ | ] |
|||||||
|
GSK3326595 |
|
Substrate competitive |
|||||||
|
NCT04676516 |
GSK3326595 |
Phase II |
6.2 nM |
Early-stage breast cancer Antitumor effect in non-Hodgkin lymphoma cell line mouse xenograft and antitumor effect in a DLBCL PDX mouse model |
|||||
|
AMG 193 |
|||||||||
|
NCT03886831 |
Structure undisclosed |
MTA cooperative inhibitor |
PRT543 |
Phase I NA |
Advanced solid tumors and hematological malignancies Antitumor effect on advanced/metastatic solid tumors |
[119] |
mTOR signaling | ||
|
PRT543 |
|
Methylation (hnRNPA1) |
SAM competitive |
10.8 nM |
Antitumor effect on advanced solid tumors and hematologic malignancies |
||||
|
NCT05275478 |
TNG908 | [ | 75] |
||||||
Phase I and II (recruiting) |
Locally advanced solid tumors | [120] |
DNA instability response |
Altered expression of RNF168 |
|||||
|
PRT382 |
|||||||||
|
NCT04089449 |
Structure undisclosed |
PRT811SAM competitive |
2.8 nM |
Antitumor effect on hematological tumors |
Phase I (recruiting) [76] |
||||
[ | ] | ||||||||
Advanced solid tumors, recurrent glioma, and CNS lymphoma |
PRT811 Cell migration, cell cycle progression, and apoptosis |
Structure undisclosed |
|||||||
|
NCT05245500 | SAM |
MRTX1719Altered expression of LRP12 |
competitive [62] |
||||||
3.9 nM |
Phase I and II (recruiting) Antitumor effect on advanced solid tumor, Glioblastoma, CNS Lymphoma |
Mesothelioma, NSCLC, malignant peripheral nerve sheath tumors, solid tumors, and pancreatic adenocarcinoma [121] |
|||||||
|
TNG908 |
AKT signaling and metastasis |
||||||||
|
NCT05094336 |
Structure undisclosed |
Methylation of PKB |
MTA cooperative inhibitor [77] |
||||||
AMG 193 | 110 nM |
Phase I and II (recruiting) Antitumor effect on Glioblastoma, |
[122] |
||||||
Advanced MTAP-null solid tumors |
Lungs |
MRTX1719 Metastasis |
|
||||||
|
NCT05528055 |
SCR6920 | Altered expression of EMT genes |
PRMT5–MTA complex inhibitor, MTA competitive [78 |
Phase I (recruiting)] |
|||||
12 nM | Antitumor effect on solid tumor |
||||||||
Advanced malignant tumors |
LLY-283 (C220) Metastasis |
Altered expression of SHARPIN |
|
SAM | |||||
|
NCT05015309 |
SH3765 |
competitive |
[79 |
Phase I (not yet Recruiting) ] |
|||||
22 nM |
Advanced malignant tumors |
Reduced acute graft versus host disease incidence in mice, antitumor effect in MPN xenografts |
|||||||
|
Compound1a |
Metastasis |
Altered expression of FGFR3/miR-99 family |
Allosteric modulator [80] |
||||||
16 nM | Antitumor effect in breast cancer |
[127] |
Metastasis |
Methylation of KLF5 |
[81] |
||||
|
CMP5 |
Structure undisclosed |
SAM competitive |
25 µM |
Antitumor effect in breast cancer and glioblastoma |
[72] |
Liver |
Lipid metabolism |
Methylation of SREBP |
[47] |
|
JBI-778 |
Structure undisclosed |
Substrate competitive |
27 to 700 nM |
Antitumor effect in glioblastoma |
[128] |
||||
|
SH3765 |
ERK signaling |
Altered expression of BTG2 |
[82] |
||||||
Structure undisclosed |
Substrate competitive |
NA |
Antitumor effect on advanced malignant tumors, including solid tumors and non-Hodgkin lymphoma |
PRMY5 deprivation |
PRMT5 activity of LINC01138 |
[83] |
|||
[ | ] | ||||||||
|
SCR6920 |
Structure undisclosed |
Substrate competitive |
NA |
Antitumor effect on advanced malignant tumor including solid tumor and non-Hodgkin lymphoma |
[129] |
WNT-β-Catenin signaling |
Altered cofactor binding of LYRIC |
[84] |
|
|
Spleen |
NA |
Altered stability of MYC |
[85] |
||||||
|
Pancreas |
Glucose metabolism, cell cycle progression |
Altered stability of MYC |
|||||||
|
Bone |
Type I interferon signaling |
Altered expression (interferon gene) |
|||||||
|
Prostate |
AR, ERG signaling |
Altered methylation (AR) |
|||||||
|
Ovary |
NA |
Altered methylation (E2F1) |
[92] |
||||||
|
Heart |
Transcriptional activity |
Methylation (GATA4) |
[45] |
||||||
|
Breast |
Stemness |
Altered expression of C-MYC, KLF4, and OCT4 |
[93] |
||||||
|
Stemness |
Altered expression of FOXP1 |
[94] |
|||||||
|
Metastasis and invasion |
Altered expression of AKT genes |
[78] |
|||||||
|
Metastasis and AKT signaling |
Methylation of AKT |
[95] |
|||||||
|
Cell cycle progression |
Methylation of KLF4 |
[96] |
|||||||
|
Cell migration |
Methylation of ZNF326 |
[97] |
|||||||
|
NA |
Methylation of PDCD4 |
[98] |
Abbreviations: AKT-ERK, alpha serine/threonine-protein-extracellular-regulated kinase; AR, androgen receptor; E2F1, E2 promoter binding factor 1; EMT, epithelial–mesenchymal transition; ERG, ETS-related gene; FGFR3, fibroblast growth factor 3; FOXP1, forkhead box protein P1; FUS, fused in sarcoma; GSK3β-NF-kβ, glycogen synthase kinase; hnRNPA1, heterogeneous nuclear ribonucleoprotein A1; HTT, huntingtin protein; KLF4, Kruppel-like factor 4; LRP12, low-density lipoprotein receptor-related protein 12; LINC01138, long non-coding RNA; miR-99, microRNA99; mTOR, mammalian target of rapamycin; OCT4, octamer binding protein 4; PKB, protein kinase B; PDCD4, program cell death protein 4; SREBP, sterol regulatory element-binding protein; ZNF326, zinc finger protein 326.
3. PRMT5 Association with MYC-Driven Medulloblastoma
Epigenetic deregulation plays a key role in medulloblastoma tumorigenesis, especially in aggressive Group 3 and Group 4 medulloblastomas [99][100][101][102][99,100,101,102], where germline mutations in known cancer predisposition genes are rare. Indeed, epigenetic deregulator or chromatin modifiers, including histone acetylase or methylation/methyltransferase activities, are very common in Group 3 and 4 medulloblastomas compared to other subgroups. This emphasizes the need to discover and understand the pertinent mechanisms of epigenetic regulation or PTMs and the corresponding therapeutic targets. ThWe researchers receecently reported that PRTM5 is a critical regulator MYC oncoprotein in an MYC-amplified (Group 3) medulloblastoma [22]. ThWe researchers found that high levels of PRMT5 not only mirror MYC expression but also correlate with poor outcomes in Group 3 medulloblastoma patients. Mechanistically, the researchers shwe showed that PRMT5 stabilizes the MYC protein by physically interacting with it, raising the intriguing possibility that PRMT5 can regulate MYC function at both the transcriptional and translational/post-translational levels. The exact MYC oncogenic programs regulated by PRMT5 in medulloblastoma are largely unknown. Therefore, exploring the regulation of MYC-driven oncogenic progresses by PRMT5 is crucial to identify effective therapeutics for these high-risk patients. The involvement of PRMT5 has been verified in the epigenetic regulation of chromatin complexes following interaction with numerous proteins, including transcription factors [42], and their activities are dysregulated in various cancers [59]. In recent studies, high levels of PRMT5 and MYC corelate with glioma malignancy [61][62][63][61,62,63]. Furthermore, PRMT5 is physically associated with N-MYC (an MYC homologue) and enhances the stability of N-MYC in neuroblastoma cells [51]. Nonetheless, the function of PRMT5 and its interaction with MYC in MYC-driven medulloblastoma have not been fully investigated. Favia et al. reported that the association of PRMT1 and PRMT5 with MYC in glioblastoma stem cells resulted in MYC being dimethylated symmetrically and asymmetrically by both enzymes, respectively [103]. MYC-driven cellular processes resulting from symmetric dimethylation by PRMT5 are shown in Figure 3. The colocalization of PRMT5 and MYC suggests that PRMT5 forms a complex with MYC and supports its stabilization in MYC-amplified medulloblastoma cells. This physical interaction of PRMT5 and MYC implies a potential role of PRMT5 in medulloblastoma tumorigenesis.