非酒精性脂肪性肝病(Nonalcoholic fatty liver disease (NAFLD)在西方国家已成为一种越来越常见的疾病,近几十年来已成为除病毒性肝炎外肝硬化或肝细胞癌(HCC)的主要原因。此外,研究表明,NAFLD与肝外疾病的发展有着千丝万缕的联系。然而,目前还没有有效的治疗方法来治愈NAFLD。此外,2020年将NAFLD更名为代谢功能障碍脂肪肝(MAFLD),以表明其发病机制与代谢紊乱密切相关。最近的研究报告说,MAFLD的发展与肝细胞和肝星状细胞(HSC)的线粒体功能障碍密不可分。同时,由结构和功能障碍引起的线粒体应激刺激肝细胞和造血干细胞中脂肪和脂肪毒性的发生和积累。此外,线粒体功能障碍与肝-肠轴的相互作用也成为MAFLD发展过程中的一个新点。在这篇综述中,我们总结了几种潜在的MAFLD治疗策略的效果,包括抗氧化剂、试剂、肠道微生物和代谢物。
) has become an increasingly common diseases in Western countries and has become the major cause of liver cirrhosis or hepatocellular carcinoma (HCC) in addition to viral hepatitis in recent decades. Furthermore, studies have shown that NAFLD is inextricably linked to the development of extrahepatic diseases. In 2020, NAFLD was renamed metabolic dysfunction fatty liver disease (MAFLD) to show that its pathogenesis is closely related to metabolic disorders. And the interaction between mitochondrial dys-function and the liver–gut axis has also become a new point during the development of MAFLD.
- MAFLD
- fatty acid metabolism
- oxidative stress
- mitochondrial quality control
- liver–gutaxis
- mitochondrial antioxidant
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is the accumulation of fatty degeneration and lipo-toxicity due to intracellular lipid overload in the liver without alcohol, which further leads to liver fibrosis and finally evolves into nonalcoholic steatohepatitis (NASH) and liver cirrhosis [1–3]. In the past decade, NAFLD has become the main cause of hepatocellular carcinoma (HCC) in addition to chronic hepatitis B (CHB) and chronic hepatitis C (CHC) and is a common chronic liver disease in Western countries [4,5]. According to statistics, the global incidence of NAFLD has reached 25%, and it is associated with a higher risk of disease under the influence of basic metabolic diseases such as obesity and type 2 diabetes (T2DM) [6–8]. In 2020, the International Expert Group issued a statement proposing to rename NAFLD to metabolic dysfunction fatty liver disease (MAFLD) and rename NASH to metabolic dysfunction-associated steatohepatitis (MASH) [9,10]. This means that its pathogenesis is closely related to metabolic disorders. However, there is currently no effective treatment that can reverse or cure MAFLD or MASH in the clinic. According to studies and reports, the pathogenesis of MAFLD is diverse and complex, and there is no complete and systematic conclusion about the pathogenesis of MAFLD.
Oxidative stress and insulin resistance are recognized as hallmarks of MAFLD [11]. At the same time, metabolic syndrome, which includes conditions such as mitochondrial dysfunction and oxidative stress, inherent immune regulation disorder, and abnormal regulation of autophagy, was found to be an important factor affecting the development of MAFLD in clinical patients [2,11–20]. For example, An, P. et al. found that the copy number of mitochondrial DNA (mt-DNA) was significantly increased in MAFLD patients [21]. Pirola, C. J. et al. and Einer, C. et al., through liver biopsy of MASH mice, revealed morphological changes such as volume expansion of hepatocellular mitochondria, rounding of cristae, enhanced fluidity of the mitochondrial membrane, and loss of typical dense mitochondrial granules. In addition, they also found that the development of MAFLD was regulated by the transcriptional activity and surface modification degree of mt-DNA [22,23]. In recent years, intestinal microorganisms have also received widespread attention [24]. Rao, Y. et al. found that intestinal microorganisms can cause lipid metabolism and electron transport chain damage by inhibiting the secretion of short-chain fatty acids (SCFAs), thereby affecting the progression of MAFLD in rodents [25].
Mitochondria are the main sites of intracellular energy generation and oxidative metabolism of carbohydrates and fatty acids in cells by affecting various physiological mechanisms in MAFLD. Therefore, we have listed serval differences in mitochondrial dysfunction in chronic liver diseases. This review focuses on the causes of mitochondrial dysfunction in MAFLD and the mechanism by which mitochondrial dysfunction affects the conversion of MAFLD to HCC. The main therapeutic strategies about mitochondrial functions for and other chronic liver diseases are summarized in Table 1.
Table 1. Mitochondrial functions in different chronic liver diseases.
|
|
Steatotic Liver Disease (SLD) [26–28] |
Metabolic Dysfunction-Associated SLD (MASLD/MAFLD) [29–34] |
Excessive Alcohol and Metabolic-Associated SLD (MetAL [35–37] |
Drug-Induced Liver Injury (DILI) [38–40] |
|
structure |
The electron transport chain (ETC) is disrupted; the activity of mitochondrial complex III is decreased. |
Mitochondrial membrane permeability increases; e disappear and giant mitochondria appear; and membrane potential decreases. |
Mitochondria swell; mitochondrial membranes rupture; and membrane potential disappears. |
Mitochondrial outer membrane is damaged; mitochondrial membrane potential decreases. |
|
Energy metabolism |
ATP synthesis is inhibited; the TCA cycle is disrupted. |
ATP synthesis is reduced; oxidative phosphorylation and fatty acid oxidation efficiency are reduced. |
ATP synthesis is downregulated. |
ATP deficiency; ETC is damaged; and succinic acid is accumulated. |
|
Mitochondrial DNA (mt-DNA) |
mt-DNA content and mitochondrial density increases. |
The fragmentation of mt- DNA is increased; the frequency of mitochondrial mutations is increased. |
The fragmentation of mt-DNA is increased. |
The copy number of mitochondrial is reduced and mt-DNA is depleted. |
|
Mitophagy |
Mitophagy is reduced. |
Mitophagy is reduced; mitochondrial quality control homeostasis is imbalanced; and mitochondrial fission is increased. |
Excessive mitochondrial autophagy. |
Mitochondrial selective autophagy is reduced. |
|
mt-ROS |
mt-ROS is increased, which is caused by incomplete oxidation of substrates such as succinic acid. |
The increase in mt-ROS production is affected by diet, lifestyle, genes, etc. |
mt-ROS increases via fat accumulation, etc. |
mt-ROS increases. |
21.Mitochondrial Structure and Function in MAFLD
21.1 Mitochondrial Membrane Structure
21.2 Mitochondrial DNA Mutation
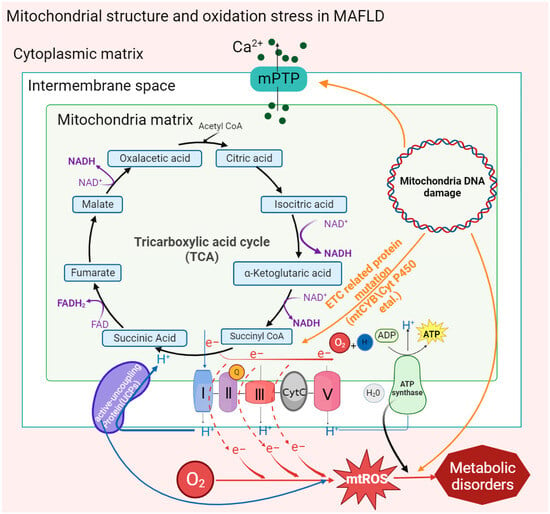
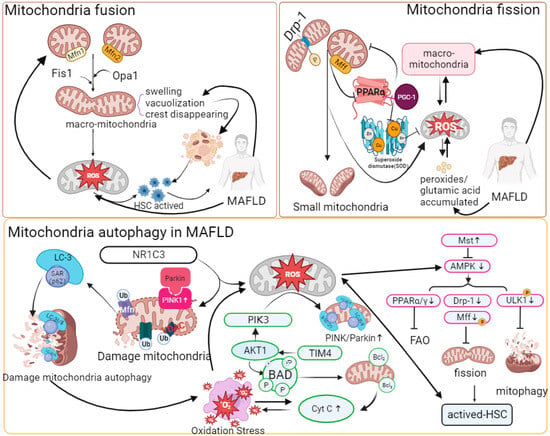
21.3 Mitochondrial Quality Control
21.4 Mitophagy
21.5 Oxidative Phosphorylation
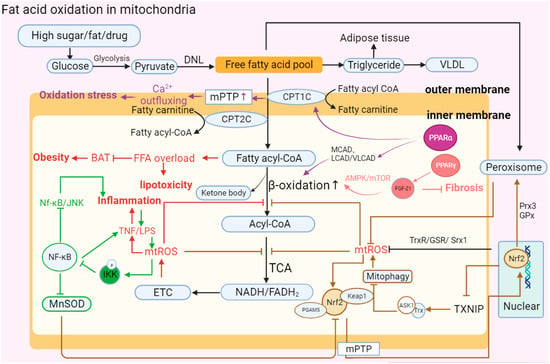
21.6 Fatty Acid Oxidation (FAO)
21.7 Gut Microbiota in the Liver–Gut Axis Influence Mitochondrial Function
3. Treatment Approaches for Mitochondrial Dysfunction-Related Oxidative Stress in MAFLD
In recent years, the prevalence of MAFLD has been increasing year by year according to the proportion of regions. Through the statistics of the current clinical research on MAFLD, it was found that there is medicine that can target metabolic processes in different ways to alleviate MAFLD.
3.1. Antioxidant Trace Elements
By comparing the serum of MAFLD and MASH patients with that of normal people, it was found that the expression of vitamins in the serum showed a significant downward trend [113,131]. According to a number of experimental models, injecting vitamins can significantly improve the occurrence of inflammation, liver fibrosis, and the worsening trend of MAFLD. The vitamin family is an important class of antioxidant molecules in the body that regulate oxidative stress and fat accumulation by catalyzing enzymatic reactions.
3.1.1. Vitamin C
Vitamin C (VC) is a common water-soluble molecule, and we often supplement the VC content in the body in the form of an aqueous solution daily. According to its antioxidant properties, VC plays an important role in scavenging oxygen free radicals. The basic mechanism by which VC alleviates the degree of oxidative stress is through reducing the generation of mt-ROS and increasing the levels of antioxidant enzymes such as superoxide dismutase and GSH peroxidase, thereby improving the activity of the ETC [83]. At the same time, VC inhibits fat accumulation by activating PPARα in MAFLD. High-dose VC stimulates the downregulation of adiponectin, which has inhibitory effects on lipid accumulation, IR, and inflammation in the liver [132].
Clinical investigations have shown that the lack of VC in the body (VC deficiency) successively causes liver fibrosis and MAFLD aggravation, especially in obese patients or overweight patients (BMI > 24) [133]. In the mouse model of a high-fat diet, appropriately supplementing a large amount of VC (beyond what is necessary for the body) would effectively alleviate the fat accumulation and MAFLD caused by high sugar and fat. At the same time, different concentrations of VC were used to treat MAFLD, and it was found that supplementation with low concentrations had positive significance in preventing liver inflammatory diseases in normal mice [132,134]. In addition, it was confirmed in a guinea pig model that VC deficiency has no significant effect on the development of MAFLD under a low-fat diet, but if a high amount of VC was combined with a low-fat diet, it obviously promoted the development of liver inflammatory disease reversal in healthy liver [135]. Even though in different animal models the intake of microbial C has different significance for the occurrence of MAFLD, the conclusion of the comprehensive experiment found that VC is indeed a good target for clinical intervention of liver inflammatory diseases. However, its specific function in the human body and therapeutic drug doses still need further investigation.
3.1.2. Vitamin E
Vitamin (VE) is an important antioxidant inhibitor against the conversion of simple steatosis to MASH caused by excessive oxidative stress. Similar to the role of VC, the detoxification function of VE is mainly through capturing and providing electrons for free hydroxyl radicals and H2O2 and then combining with antioxidant enzymes to detoxify them into water and oxygen [136]. VE also has potential effects on the development of inflammation. VE downregulates the expression of trans growth factor-β (TGF-β) through mt-ROS [136], resulting in reduced forms of nitric oxide synthase and NADPH oxidase by reducing the frequency of oxidative stress to affect the development of liver fibrosis [137]. In addition, it was found in hepatitis mice that feeding with an MCD diet can restore glutathione and significantly reduce the expression levels of oxidative stress markers, HSC activation, and fibrosis under VE injection [138,139]. VE can also activate PPARα transcription, promote the action of the adiponectin promoter, and enhance IR, thereby alleviating the development of MAFLD [140].
Due to the active physiological functions of VE, it is also recognized as an effective targeted therapy drug in clinical treatment. In data collected through broad-spectrum surveys of the population, it was found that VE tended to show more obvious light fibrosis symptoms than control groups [141]. In addition, the results showed that in children with nonalcoholic liver disease, compared with the injection of a placebo, hydroxytyrosol and vitamin E significantly improved the degree of steatosis and weakened the occurrence of systemic inflammation by promoting the systemic circulation of the anti-inflammatory factor interleukin-10 [13]. In conclusion, VE is widely used in combination with other biological drugs to regulate the level of hepatic oxidative stress and FAO, but its specific appropriate doses for the clinical treatment of MAFLD and MASH still need to be further determined and explored for clinical applications.
3.1.3. Vitamin D
Vitamin D (VD) is a multifunctional hormone that not only stabilizes calcium homeostasis and regulates bone mineralization but also plays an important role in the regulation of immunity and inflammation. VD deficiency has been associated with the development of metabolic syndrome-associated diseases such as T2DM and MAFLD [142]. In a study of FFA decomposition in MAFLD models, VD acted on outer membrane lipoprotein through the PPARα/CPT1A pathway to catalyze the mitochondrial β-oxidation process in hepatocytes and inhibited the denaturation of lipoproteins and reduced the development of MAFLD [143].
VD also controls the endoplasmic reticulum–mitochondrial stress-oxidative response by promoting the nuclear translocation of the antioxidant molecule nuclear factor erythroid 2-associated factor 2 (NFE2L2), reducing Toll-like receptor expression [144,145]. In addition, a study showed that VD mediated the activation of hepatocyte nuclear factor 4α (HNF4α) through specific receptors to improve hepatic insulin resistance and reduce the possibility of hepatic steatosis [143] At present, the effect of VD on the clinical treatment of chronic inflammatory liver diseases is still unstable, and the impacts of the intake of different concentrations of VD on health are worthy of further research.
3.1.4. Vitamin A
Vitamin A (VA) is a necessary regulatory factor for many physiological processes, such as visual perception, cell proliferation and differentiation, immune response, and metabolic regulation. Retinoic acid (retinol), a metabolite of vitamin A, plays an important role in the function of VA. Retinoic acid receptor (RAR) and retinoid receptor (RXR) jointly control the activation of retinoic acid transcription factors. The RAR and RXR receptors, which are the unique structure of vitamin A, can also affect the process of fat oxidation by acting on the PPARα pathway [146].
Nonactivated HSCs in the liver are the main storage location of VA in the body, which also reveals that VA deficiency is inextricably linked to the occurrence of liver fibrosis [147]. According to the data collected in existing clinical research from MAFLD patients, VA metabolism disorder is not only manifested in the downregulation of overall expression in serum but also shows an imbalance in the ratio of retinal lipids and retinol, as well as a change in intracellular retinoid storage locations, such as retinol transferred from HSCs to hepatocytes [103]. However, summarizing recent research, it has been found that the treatment of chronic liver inflammatory diseases by directly increasing vitamin A intake is not stable for the treatment of MAFLD. This may be related to the individual differences in patients and related molecular mechanisms, such as retinoid lipid overload accumulation in hepatocytes, which also provides a new direction for further exploration of VA to improve HSC activation and FFA accumulation in MAFLD.
3.1.5. Vitamin B
In the past decade, several vitamin B subfamilies have been studied in MAFLD conditions in existing clinical studies.
Vitamin B3 (Niacin) is the precursor molecule of the coenzymes nicotinamide adenine dinucleotide (NAD) and nicotinamide adenine dinucleotide phosphate (NADPH), which regulate various physiological responses [133,148]. In a rat model and a high-fat HepG2 model, increasing niacin restored the loss of mitochondrial redox potential, ROS accumulation, lipid digestion in the liver, and NADPH enzyme activity reduction in chronic inflammation [149,150].
Vitamin B9 (folic acid) and vitamin B12 (cyanocobalamin) are both related to the occurrence of MAFLD-related comorbidities such as obesity and T2DM. Folate acid protects the liver by restoring activation of adenosine monophosphate-activated protein kinase (AMPK). Vitamin B12 is the co-factor of methylmalonyl-CoA mutant enzymes that affect DNA synthesis repair and mitochondrial metabolic homeostasis, which are involved in the pathogenesis of MAFLD [151]. However, according to current clinical case data, there are not enough complete and sufficient results to explain whether the injection of the vitamin B family regulates the occurrence of MAFLD [152,153].
3.1.6. Coenzyme Q
Coenzyme Q (CoQ) is a self-synthesized fat-soluble bioactive quinone, similar to vitamin E. CoQ is located on the inner membrane of mitochondria and is widely distributed in various tissue cells. The function of CoQ is mainly through participating in mitochondrial ETM, improving electron transport efficiency, and maintaining redox homeostasis by utilizing the structural transformation between the three redox forms of ubiquinone, semi-ubiquinone, and ubiquinol, and directly acting on free radicals and oxides to restore cell viability [154].
Clinical data have shown that CoQ10 supplements can alleviate the oxidation level of type I, II, and III complexes in the respiratory chain, regulating OXPHOS and inhibiting mitochondrial dysfunction induced by MAFLD [155]. Alhusaini, A. M. et al. found that liposome-encapsulated coenzyme Q could significantly reduce liver damage and fibrosis caused by propionic acid by inhibiting cytochrome C and mitochondrial fragmentation and simultaneously increasing the expression of Bcl-2 [156]. Tiefenbach, J. et al. proposed that coenzyme Q and its analog Idebenone can act as PPARα/γ agonists and downregulate triglyceride and cholesterol levels, finally reducing the development of steatosis and MAFLD [157]. Sumbalova, Z. et al. used hydrogen-rich water (HRW) to treat MAFLD clinical patients and mouse models and found that HRW has the potential to help MAFLD patients restore normal coenzyme Q expression levels and mitochondrial oxidation function, ultimately realizing the potential of MAFLD treatment [158]. In mouse MAFLD models, very-low-density lipoprotein (VLDL) was overproduced in a high-fat diet. CoQ10 was shown to guide VLDL to accumulate and transform into a larger volume so that it is more easily recognized and degraded by enzymes to control the occurrence of lipid peroxidation and oxidative stress [159]. Notably, a large volume of VLDL also causes other vascular diseases, such as arteriosclerosis and other side effects. Hence, it is important to solve the main question about the by-effect in the process of treating MAFLD for CoQ through clinical treatment or animal models.
3.2. Nrf2Antioxidant Supplement
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a high-affinity electron-competent transcription factor involved in antioxidant effects. Nrf2 can inhibit excessive oxidative reactions such as endoplasmic reticulum stress by regulating downstream antioxidative stress genes such as heme oxygenase-1 (HO-1) and superoxide dismutase (SOD). Due to its outstanding antioxidant capacity, there are currently a variety of Nrf2 supplements used in the disease research of MAFLD and MASH [87].
3.2.1. Aucubin
Aucubin (AU) is a natural compound that can be extracted from plants and has anti-inflammatory effects. AU relieves lipid accumulation by promoting the expression of Nrf2 and PPAR in mice and 3T3-L1 cells (a type of macrophage) and inhibits the release of proinflammatory cytokines such as TNF-α, IL-1β, and IL-6, in turn reducing oxidative stress and the inflammatory response by enhancing the oxidative stress and AMPK/AKT phosphorylation associated with hyperlipidemia [160]. AU could counteract the hepatic fibrosis caused by CCL4 and α-amanitin, showing therapeutic significance for the treatment of MAFLD.
3.2.2. Melatonin
Melatonin is a small molecule indole amine substance that is mainly related to the regulation of biological rhythm in animals. Melatonin has antioxidant properties and counteracts the negative effects of active oxygen in the body. Joshi, A. et al. found that melatonin-mediated HepG2 cells reduce oleic acid uptake and increase mitochondrial membrane potential. In high-fat-diet-fed mice treated with melatonin, Nrf2/HO-1 activity was restored, and the expression of Nrf2-Keap1 in hepatocellular mitochondria was increased, reducing intracellular oxidative stress levels and alleviating MAFLD [161].
Research on drugs targeting the Nrf2/OH-1/Keap1 pathway, intracellular oxidation reactions, and lipid degradation processes has become a new hot topic. In addition to AU and melatonin, a variety of natural compounds have been used in the prevention of MAFLD/MASH and have obvious treatment effects. For example, inhibition of the upregulation of p62 transcription regulated by ROS/P38/Nrf2 alleviated the oxidative stress damage of macrophages caused by LPS [58,162]. Naringin downregulates the expression of the targeted proteins ChREBP, SREBP-1c, nSREBP-1c, ACC, and FAS to inhibit the accumulation of fatty acids and regulate Nrf2-HO-1/Nf-κB to reduce the impact of inflammation on the development of MAFLD [163].
3.3. MicroRNA
MicroRNA (miRNA), as an important bioactive factor target, has been widely found and studied in specific epigenetic mechanisms. In recent years, abnormal expression of miRNAs has been found in metabolic disorders such as MAFLD and MASH [164–166]. For example, it was found that miRNA-21/20B was upregulated in the inflammatory environment in the HFD-fed mouse model. Mt-ROS and nitric oxide (NO) generated from activated HSCs could target miRNA-21 to form more pro-fibrotic proteins, such as type I collagen (Col1α1) and α-SMA [136]. MiRNA20B in MAFLD mediates PPAR-α activity to reduce the occurrence of abnormal FFA oxidation and mitochondrial dysfunction [167]. In addition, circRNA-002581 inhibits the expression of miRNA-122, which promotes fat oxidation and the activation and phosphorylation of mTOR to aggravate the development of inflammation [168].
Other studies confirmed that miRNA-34a and miRNA-223 inhibited the Sirtuin 1/AMPK/PPARα pathway through vesicle enrichment [169,170]. MiRNAs play a therapeutic role in the process of chronic inflammation and oxidative stress. As a result, miRNAs have become new treatments to eliminate the development of MAFLD and MASH.
3.4. Targeted Microbiota Therapies Targeting the Liver–Gut Axis
3.4.1. Gut Myxiniphila
In recent years, beneficial microorganisms such as Akkermansia muciniphila have been considered promising research hotspots in combating MAFLD. Morrison, M. C. et al. used heated Myxiniphila to treat HFD-induced obese mice and found that it could significantly decrease intestinal permeability and adipocyte hypertrophy [171]. Rao, Y. et al. treated MAFLD mice with oral administration of Proteobacillus Myxophila and found that mt-DNA copy number and oxidative metabolism markers such as PGC-1α and CPT-1β increased in the hepatocytes of the treatment group. In addition, the expression of mitochondrial complexes I–V was significantly upregulated. In addition, Myxophila stimulated L-aspartic acid, thereby activating AMPK activity and sustaining lipid oxidation reactions, effectively inhibiting liver lipid accumulation [25]. At this stage, various research data show that whether it is through direct oral administration or through dietary or drug intervention in the MAFLD mouse model, the abundance of Myxophila in the body shows great therapeutic potential for the alleviation of MAFLD. However, clinical efficacy verification in other animals is still lacking, and the reduction in therapeutic efficacy after inactivation needs further investigation [172,173].
3.4.2. Bile Acids (BAs) and Short-Chain Fatty Acids (SCFAs)
Intestinal metabolites such as bile acids (BAs) and short-chain fatty acids (SCFAs) are important molecules involved in the enterohepatic circulation and metabolism of the body, affecting the development of MAFLD. BAs regulate lipid metabolism, gluconeogenesis, and ATP synthesis processes by activating Farnesoid X receptor (FXR), the G protein-coupled receptor superfamily (TGR5), and other receptors that are highly expressed in the liver and intestines [174]. The FXR-TGR5 dual agonist INT-767-treated Western diet (WD)-fed mice could reduce fatty acid synthesis and alleviate AMPK, SIRT1/SIRT3 phosphorylation, and mitochondrial complex IV activity, decreasing trends caused by WD [175]. However, excessive activation of FXR can lead to an increase in total cholesterol and low-density lipoprotein cholesterol levels in MASH patients; therefore, the safe dosage of FXR agonists still needs further prediction and evaluation.
Short-chain fatty acids (SCFAs) are a class of saturated and fatty acids produced by intestinal flora through fermentation of soluble dietary fiber, including acetate, propionate, and butyrate. Zhao, T. et al. found that butyrate supplementation could inhibit the activity of NADH oxidase in the ETC of MAFLD patients, increase the concentration of potassium ions in the mitochondria, maintain the mitochondrial membrane potential, and delay the development of MAFLD [130,176]. Acetate and propionate mainly maintain the intestinal barrier and reduce the release of inflammatory factors such as IL-6 [177].
3.5. Other Molecular Drugs
Several experimental targets are being explored in clinical practice to delay the sustained development of MASH, including FXR agonists, FGF21, glucagon-like peptide-1 (GLP-1), PPAR agonists, etc. GLP-1 is an incretin hormone that regulates blood sugar and weight homeostasis. Clinical diabetic patients with low expression of GLP-1 receptor (GLP-1R) are known to have mt-ROS accumulation, superoxide formation, and membrane potential loss [178]. The GLP-1 analogs liraglutide and semaglutide have been proven to delay the development of MASH. Semaglutide can reduce the occurrence of mitochondrial dysfunction by enhancing autophagy and resisting oxidative stress in neurodegenerative diseases [179]]. During the treatment of MASH patients, semaglutide can also bind with GLP-1R to effectively improve glucose–lipid metabolism and reduce oxidative stress in hepatocytes [180]. Liraglutide increases the levels of mitochondria-related structural proteins, such as Drp1, OPA1, and UCP2, enhances mitochondrial structural remodeling, and restores the expression of autophagy-related proteins, such as Beclin 1, LC 3, and Sirtuin 1, to inhibit the development of MAFLD [181]. The combined use of multiple drugs has shown a stronger effect on the control of MAFLD than single drugs. For example, the combined use of FXR agonist (Cilofexor), ACC inhibitor (Firsocostat), and semaglutide in MASH patients was compared with semaglutide alone, and the combined use of the drugs improved the two indicators of transaminase and fat content more significantly [182] (Table 2).
Table 2. The main therapeutic strategies for mitochondrial function in MAFLD/MASH.
|
Name |
Pathway |
Treatment |
Model |
Effect for MAFLD |
|
Antioxidant trace elements |
||||
|
Vitamin C (VC) |
mt-ROS↓, adiponectin↓ PPARα↑, antioxidant enzymes↑ |
High-dose intake |
Mouse (High-fat diet) |
Reduce lipid accumulation, IR, and inflammation |
|
High-dose intake + low-fat diet |
Guinea pig |
|||
|
Vitamin E (VE) |
Mt-ROS↓, iNOS↓, NADPH oxidase↓; PPARα↑ |
Oral administration of hydroxytyrosol and VE |
MAFLD children |
Reduce HSC activation and fibrosis |
|
Vitamin D (VD) |
mTOR↓, Sirtuin↓; PPARα/CPT1A↑, HNF4α↑, NFE2L2↑ |
Gavage dose of VD |
Wistar rats |
Reduce liver steatosis, serum lipid accumulation |
|
VD |
HepG2 cell by OA |
Inhibit lipid and TG accumulation in cell |
||
|
Vitamin A (VA) |
PPARα↑, FGF21↑, CPT1A↑, UCP2↑ |
—— |
—— |
VA deficiency in MAFLD patients |
|
Vitamin B (VB) |
ROS↓; restore lipid digestion, the activity of NADPH enzyme, and mitochondrial redox potential |
0.5% niacin in the diet |
Rat (High-fat diet) |
Reduce chronic inflammation and hepatic steatosis |
|
Coenzyme Q |
||||
|
L-辅酶Q101 |
恢复OXPHOS和线粒体自噬;增加细胞色素C的活性 |
Oral administration L-辅酶Q10 |
Rat (口服PRA诱导的MAH中毒) |
减少肝损伤和纤维化 |
|
艾地苯醌 |
PPARα/γ↑;甘油三酯↓、胆固醇↓ |
口服艾地苯醌 |
2型糖尿病小鼠模型 |
减少肝脂肪变性 |
|
富氢水 |
恢复正常的辅酶 Q 表达水平 |
口服 |
MAFLDH患者/小鼠模型 |
缓解 MAFLD 的潜在疗法 |
|
Nrf2 抗氧化剂补充剂 |
||||
|
奥库宾 |
Nrf2↑, PPAR↑, p-AMPK/AKT↑;TNF-α↓、IL-1β↓、IL-6↓ |
腹腔注射aucubin |
鼠 (泰洛索诱导的MAFLD) |
减少脂质积累、氧化应激和炎症 |
|
褪 黑 素 |
MT-ROS↓;恢复 Nrf2/HO-1 和线粒体氧化还原电位的活性 |
腹腔注射褪黑激素 |
鼠 (高脂肪饮食) |
减少氧化应激和对肝细胞的损伤 |
|
—— |
HepG2 的 OA |
|||
|
东莨菪酮 |
MT-ROS/P38/NRF2↓、P62↓ |
—— |
LPS的巨噬细胞 |
缓解氧化应激损伤 |
|
微核糖核酸 |
||||
|
miRNA21/20B |
—— |
—— |
鼠 (CCL4诱导的MAFLD) |
MASH小鼠的上调 |
|
miRNA-223(米核糖核酸-223) |
囊泡富集抑制 Sirtuin 1 和 AMPK 活化;MT-ROS↓;Nrf2↑, HO-1↑, SOD1/2↑ |
miR-223 由升高的 EA 表达 |
HepG2细胞 (高糖诱导的MAFLD) |
减少氧化应激和胰岛素抵抗 |
|
微生物群和肠道代谢物 |
||||
|
Akkermansia muciniphila 粘液菌 |
恢复mt-DNA、PGC-1α、CPT-1β,激活AMPK活性 |
口服Akkermansia muciniphila |
鼠 (HFD饮食) |
减少肝脂质堆积 |
|
胆汁酸 |
激活法尼醇 X 受体 (FXR)、G 蛋白偶联受体超家族 (TGR5) 和 ATP 合成过程 |
腹腔注射INT-767 |
鼠 (HFD饮食) |
减少脂肪酸合成、AMPK、SIRT1/SIRT3磷酸化,线粒体功能下降趋势 |
|
短链脂肪酸 |
维持线粒体膜电位 |
丁酸盐补充剂 |
MAFLD患者 |
延缓MAFLD的发展 |
|
其他分子药物和策略 |
||||
|
Semaglutide |
Bind with GLP-1R to decrease the accumulation of mt-ROS, superoxide formation, and membrane potential loss |
—— |
MASH patients |
Improve IR and glucose–lipid metabolism, and reduce oxidative stress |
|
SH-SY5Y cell |
||||
|
Liraglutide |
Drp1↑, OPA1↑, UCP2↑, Beclin1↑, LC3↑ |
Subcutaneous injection of liraglutide |
Mouse (HFD diet) |
Inhibit the development of MAFLD |
|
|
||||
“↑” represents upregulated expression; “↓” represents downregulated CoQ10(Liposomal-coenzyme Q10); SCFAs (short-chain fatty acids); PPARα (proliferator-activated receptor-alpha); FGF21 (fibroblast growth factor 21); CPT1(Carnitine Palmitoyl Transferase I); UCP2(Uncoupling Protein 2); NFE2L2 (Nuclear Factor Erythroid 2-associated Factor 2); HNF4α (Hepatocyte Nuclear Factor 4α); HFD-diet (high-fat diet); EA (ellagic acid); INT-767(FXR-TGR5 dual agonist).
4.Summary
At present, MAFLD has become a global health and safety issue by affecting a quarter of the population in the world and has no approved drug therapy. Furthermore, in recent years, studies have shown that MAFLD is inextricably linked to the development of extrahepatic diseases. Tarantino, G. et al. used the triglyceride/glucose (TyG) index and triglyceride/high-density lipoprotein (HDL) to detect the occurrence of MAFLD and IR, respectively, in bladder cancer patients and found that both TyG and HDL significantly increased [183]. Liang, Y. et al. used 6873 Chinese middle-aged and elderly people as clinical research subjects and found that patients with MAFLD had a significant increase in the risk of cardiovascular diseases by inducing abnormal elevation of arterial hyperlipidemia and causing myocardial damage [184,185]. At the same time, liver fibrosis inhibits the filtering function of glomeruli to injure kidney functions [186]. In addition, for MAFLD patients of different sexes, the risk of colon cancer in men and breast cancer in women was also significantly increased. Mantovani et al. conducted a systematic summary of the association between MAFLD and extrahepatic cancers through follow-up observation statistics of clinical patients and found that MAFLD increased the risk of extrahepatic cancer, including increasing the risk of developing gastrointestinal cancers, such as esophageal, gastric, pancreatic, and colorectal cancers, by nearly 1.5- to 2-fold [186–188]. In addition, MAFLD can also significantly increase the probability of breast cancer, thyroid disease, etc. [189]. However, it is worth noting that the probability of extrahepatic cancers increased by MAFLD is affected by a variety of confounding factors, including age, sex, smoking, obesity, and other potential factors, so these related mechanisms need to be further researched.
As important organs for energy formation and metabolism in cells, mitochondria mainly metabolize carbohydrates and fatty acids through oxidative phosphorylation and β-oxidation, respectively, in hepatocytes. However, when the mitochondrial structure is damaged, the genome expression is abnormal, or the quality control is disordered, which leads to the collapse of ETC, the reduction in ATP synthesis, and the massive accumulation of mt-ROS and intermediate metabolites of fatty acid decomposition such as dicarboxylic acid. This review mainly focuses on the causes and impacts of mitochondrial dysfunction on the pathogenesis of MAFLD. In addition, there are potential therapeutic effects of natural antioxidant compounds and molecular medicine to treat MAFLD.
In addition, Fu, A. et al. used tail vein injection of healthy mitochondria into MAFLD mice to compensate for the mitochondrial physiological activity of missing functions. This approach effectively reduced serum aminotransferase activity and blood lipid content and significantly restored ATP synthesis, cytochrome oxidase activity, and mitochondrial antioxidant formation [163]. At present, there is still a lack of other animal models or clinical cases to further determine the therapeutic effect of mitochondrial reinfusion, while it is undeniable that it will be a new potential strategy for the treatment of MAFLD.
The pathogenesis of MAFLD or MASH is highly heterogeneous and influenced by different triggers at different ages. For example, simple steatosis induced by genetics, malnutrition, or changes in the environment eventually leads to MAFLD, which is the main cause of illness in children and adolescents. In adults, there are many causes, including non-excessive alcohol, genetics, diabetes, etc. These causes can also trigger mitochondrial dysfunction. In addition, the manifestations of mitochondrial dysfunction vary among different disease types. Comprehensive clinical data analysis revealed that the number of mitochondria in the liver of patients with MASH is reduced, and the structure is more obviously damaged than that in normal people, which in turn aggravates the process of liver cell necrosis and fibrosis.
Clinical data and research provide evidence to support that mitochondrial dysfunction influences the development of MAFLD by changing oxidation reactions and antioxidant mechanisms. We have provided a simple table to summarize these main therapeutic strategies for mitochondrial function in MAFLD or MASH in this review. However, mitochondrial dysfunction in MAFLD has yet to be completely elucidated. Therefore, there is virtual significance to further determine different types of antioxidant molecules in the occurrence of MAFLD. It is equally important to determine whether the use of the above antioxidants would cause side effects and impact the treatment effect at different stages of MAFLD or MASH. Risk assessment and prediction in clinical treatments for MAFLD and MASH patients through transplantation of beneficial intestinal flora and mitochondrial reinfusion also require further verification.