Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Avisek Majumder.
It is well recognized now that the development of drug resistance is one of the leading causes of treatment failure in conventional therapies. In comparison, improvements in immunotherapy showed promising results in eradicating cancer. Chimeric antigen receptor (CAR)-T-cell therapy is one of the cancer immunotherapies that uses patient’s T cells and genetically modifies them to target cancer cells. Although CAR-T-cell therapy has shown remarkable success in treating blood cancer, it has proven far more limited in the treatment of solid tumors in different organs.
- immunotherapy
- immuno-oncology
- combination therapy
- cancer treatment
- tumor microenvironment (TME)
- chimeric antigen receptor (CAR)
- checkpoint blockade
- immunomodulation
1. Introduction
Cancer is the primary health concern and leading cause of death worldwide. The American Cancer Society estimated that 1,958,310 new cancer cases and 609,820 cancer deaths occurred in the year 2022 in the United States [1]. Although there has been enormous progress in different types of conventional therapies for treating cancer, the development of drug resistance and drug-related toxicity makes these therapies much less effective. On the other hand, immunotherapy relies on using patients’ immune systems to identify cancer cells as foreign bodies and eradicate them via various mechanisms. Various immunotherapies have been developed for cancer, primarily by potentiating the body’s immune cells by releasing their immune suppression or empowering them to more effectively perform their immune functions. Chimeric antigen receptor (CAR)-T-cell therapy is one of the approaches of adoptive T-cell transfer (ACT) used in immunotherapy, which recently showed enormous success in terms of effectiveness and durable clinical response [2]. In this approach, T cells have been genetically altered by expressing the chimeric antigen receptor (CAR) to recognize tumor-specific antigens without the involvement of major histocompatibility complex (MHC), resulting in vigorous T-cell activation and robust anti-tumor responses [3]. CAR-T cells mediate anti-tumor effects through granzyme release, cytokine release, and other immune effectors (Figure 1).
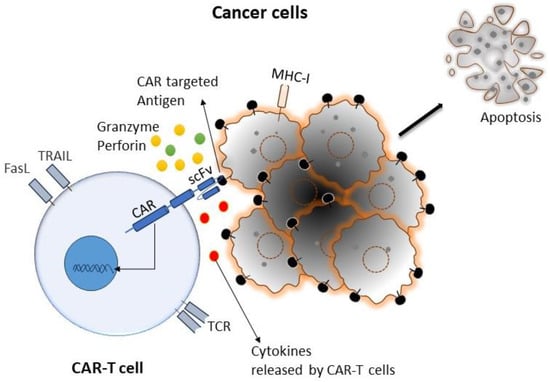
Figure 1. Cartoon diagram representing a typical interaction mechanism of CAR-T cells with targeted cancer cells: CAR-T cells can recognize tumor cells by binding with specific tumor-associated antigens (TAA) independent of TCR-MHC/peptide interactions. As a result, T cells are activated. CAR-T cells can induce apoptosis via the secretion of perforin, granzymes, and different pro-inflammatory cytokines, as well as the expression of the Fas ligand (FasL) and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL).
Growing and evolving research has utilized sophisticated ex vivo culture and cellular engineering approaches to improve CAR-T-cell therapy, with long-life therapeutic responses. As a consequence of unparalleled clinical efficacy in particular B-cell malignancies, the US Food and Drug Administration (FDA) approved many CAR-T-cell therapies for treating hematological malignancy. CAR-T-cell therapy showed remarkable efficacy and profound therapeutic potencies in treating a subset of hematological malignancies by targeting lineage-restricted surface molecules [4,5,6,7,8,9][4][5][6][7][8][9]. In contrast, CAR-T-cell therapy hits roadblocks in treating solid tumors, mainly due to lack of high-quality antigen targets and poor tumor infiltration [10]. So far, most antigens targeted in solid tumors via CAR-T cells are mainly tumor-associated antigens rather than tumor-specific ones, which means that these antigens express higher levels in tumors and lower levels in some of the normal healthy tissues. These low antigen expression in normal tissue often leads to severe on-target/off-tumor toxicities. A case report showed severe side effects on a patient’s lungs after the infusion of CAR-T cells targeting HER2 antigen, which finally leads to death after a few days [11]. CAR-T cells’ clinical success also depends on CAR-T cells’ persistence; in this regard, CAR-T cells that exhibit a long-lived memory phenotype have shown much more antitumor responses than terminal effector-like CAR-T cells [12]. Antigen-independent tonic signaling is one of the main factors affecting CAR-T cells’ phenotype and longevity, there by its therapeutic response [13]. Many studies have tested innovative techniques to overcome these limitations, including targeting dual antigens, post-translational modified tumor-associated antigens, combination therapies to reduce immunosuppressive TME, and improved penetrations to the tumor site.
2. Biology of CAR-T Cells in Cancer
Chimeric antigen receptors (CARs) are engineered to repurpose them to recognize specific antigens on the cancer cell’s surface. CAR contains a binding moiety in the extracellular part that recognizes and binds to target antigens in cancer cells and signaling domain(s) in the intracellular part that activates the T cells to proliferate and attack cancer cells (Figure 2). The binding moiety has a specific region called a single-chain variable fragment (scFv) that identifies specific antigens. This scFv region is derived from variable heavy (VH) and light (VL) chains of monoclonal antibodies connected via a flexible linker, as shown in Figure 2A. In comparison, the intracellular domain of CAR consists of several parts, which include a CD3ζ signaling domain (that activates the T cell) and co-stimulatory domains (that provide additional signals) [14,15,16,17,18][14][15][16][17][18]. Between the extra- and intra-cellular domains, there is a hinge region that provides flexibility to the extracellular domains (ECD) to help them to better recognize antigens and the transmembrane domain that anchors the CAR in the T-cell membrane.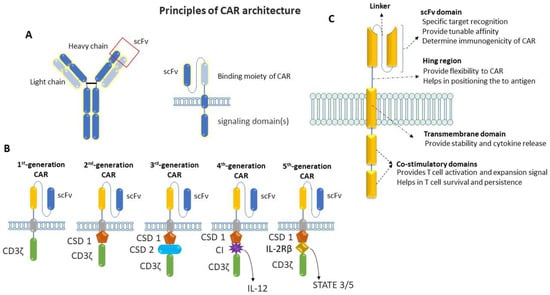
Figure 2. Schematic representation of the basic principle of CAR architecture. (A) Cartoon diagram showing the binding moiety of CAR, where scFv comes from a TAA-specific monoclonal antibody and the signaling domain(s) comes from activating and co-stimulatory immune receptors. (B) Cartoon diagram showing the progressive evolution of CAR-T cells from first generation to fifth generation: first-generation CAR contains a single-chain variable fragment (scFv) and CD3ζ for signal transduction; an additional co-stimulatory domain (co-stimulatory domain 1: CSD 1) was added on first-generation CAR to make a second generation, whereas two additional co-stimulatory domains (CSD1 and CSD 2) were added on first-generation CAR to make third-generation CAR; one additional domain for cytokine-expression (cytokine inducer: CI) was added on second-generation CAR to make fourth-generation CAR; fifth-generation CAR contains a co-stimulatory domain (co-stimulatory domain 1) and an additional domain that activates signaling pathways such as STATE3/5. (C) Cartoon diagram showing the basic structure of a typical CAR with the functions of individual domains.
3. Generation of CAR-T Cells
The first step in CAR-T-cell therapy is to collect T cells from the patient (autologous) or a donor (allogenic). Then, these T cells need to be purified and genetically engineered to express artificially generated CARs, as shown in Figure 3 [26]. An engineered CAR gene is transduced in T cells via different methods, which include viral (lenti/retrovirus), non-viral (transposon or CRISPR/Cas9), and electroporation methods [27,28,29,30][27][28][29][30]. Next, to produce a large quantity of engineered CAR-T cells, these cells were expanded in vitro. After achieving the desired quantity and quality, they were infused back into the patient’s bloodstream. Generally, patients undergo chemotherapy before the infusion of CAR-T cells into the blood stream, which allow engineered T-cells to grow and kill cancer cells. When CAR-T cells enter the patient’s bloodstream, they reach the cancer site, recognize specific antigens via CAR, and start performing their function, as detailed in the subsequent paragraphs and illustrated in Figure 1 [29].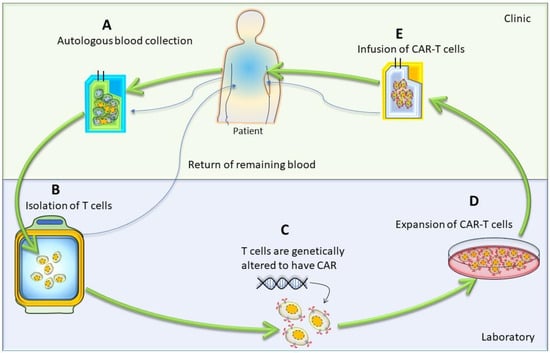
Figure 3. Cartoon diagram showing different stages of autologous CAR-T-cell production: The generation of autologous CAR-T cells started with the leukapheresis of a patient (A), followed by the separation of T cells (B); then, T cells were transduced to permanently integrate CAR transgene (C). After that, these genetically modified CAR-T cells expanded (D) and infused back into the patient (E).
4. CAR-T-Cell Therapy: From Scientific Discovery to Cures
Although CAR-T-cell therapy has recently gained much attention due to its revolutionary success in treating blood cancer, the idea has existed for several decades. It was repurposing the T-cell via genetically engineered T-cell receptors (TCRs) to kill cancer cells for the first time that was proposed in the 1980s. This approach had limitations because, at that time, it was challenging to identify specific antigens specific to cancer and make TCR specific to that antigen. This approach was even more challenging since TCR recognizes antigens in the major histocompatibility complex (MHC) setting, which may vary from patient to patient. To overcome these challenges, in 1989, it was proposed to combine the antigen-binding domain (derived from a monoclonal antibody) with the signaling domains of TCRs. This was the first generation of CAR-T cells; this approach allows the T cells to recognize the specific antigen present in cancer cells independently of MHC [31]. Although this first-generation CAR-T-cell therapy approach is more versatile than TCR, these cells have limited tumor-killing capacity because they do not grow and persist near tumors [32]. These limitations make the first generation of CAR-T cells less out of reach to the expectations of treating cancer. In the mid-1990s, the second generation of CAR-T cells was developed by adding the co-stimulatory domain to the CAR, which helps these cells to expand and survive. This approach has shown to be more efficient in preclinical models and paved the way for clinical trials. In 2002, the first effective CAR-T cells were developed by adding CD28 as a co-stimulatory domain [33]. In 2003, a study showed the complete abolition of B-cell lymphoma in a mouse model via CAR-T cells targeting the B-cell antigen CD19 [34]. In 2006, the first clinical trial was conducted with CAR targeting the tumor antigen carbonic anhydrase IX in patients with metastatic renal cell carcinoma [35]. Although this therapeutic strategy was reported to be safe for the patients, it did not yield any clinical benefits for the patients. After this step, in 2011, another clinical trial was conducted at the University of Pennsylvania, where they found clinical success in treating advanced chronic lymphocytic leukemia (CLL) using CAR-T-cell therapy [36]. This group used autologous T-cells engineered to express a CAR targeting the B-cell antigen CD19, and all patients were found to achieve complete remission after this therapy. This success led to many clinical trials with CAR-T-cell therapies, and they found various degrees of success. In 2013, a clinical trial with CAR-T-cell therapy found profound antitumor effects in patients with relapsed or refractory acute lymphoblastic leukemia (ALL) [37]. After that, in 2014, a group developed CAR-T-cell therapy for solid tumors to recognize antigen mesothelin [38]. Shortly after that, in 2015, the fourth generation of CAR-T cells was developed by adding a transgene with the second generation of CAR. After the CRISPR system was developed, this gene-editing tool was used to make CAR-T cells in 2017 [39]. Seeing the unprecedented success of CAR-T-cell therapy, in the year 2017, the US Food and Drug Administration (FDA) approved the CAR-T-cell therapy tisagenlecleucel (Kymriah) for the treatment of relapsed or refractory ALL [40]. Consequently, the FDA approved axicabtagene ciloleucel (Yescarta), another CAR-T-cell therapy for treating relapsed or refractory diffuse large B-cell lymphoma (DLBCL) in adults [40]. After that, one by one, several CAR-T-cell therapies were approved for treating different types of cancer. Some important CAR-T-cell therapies include Abecma (idecabtagene vicleucel) for the treatment of relapsed or refractory multiple myeloma [41], Lisocabtagene maraleucel (Breyanzi) for the treatment of relapsed or refractory large B-cell lymphoma [42], Brexucabtagene autoleucel (Tecartus) for the treatment of relapsed or refractory mantle cell lymphoma [43], and Liso-cel (liso-cel) for the treatment of relapsed or refractory large B-cell lymphoma [44]. The FDA approval of CAR-T-cell therapy also expanded to other types of cancer beyond leukemia and lymphoma, and there have been many ongoing clinical trials, with some being completed, which are summarized in Table 1.Table 1.
Important clinical trials for CAR-T-cell therapy in cancer that are currently ongoing or completed.
| NCT Number | CAR-T Strategy | Conditions | Type of Tumor | Phase | Current Status | Enrollment | Estimated/Actual Completion Date (DD Month YYYY) |
Sponsor |
|---|---|---|---|---|---|---|---|---|
| NCT02348216 | CD19-specific CAR-T cells | Diffuse large B-cell lymphoma (DLBCL), primary mediastinal large B-cell lymphoma (PMBCL), transformation follicular lymphoma (TFL), high-grade B-cell lymphoma (HGBCL) | Hematological malignancy | Phase 1 Phase 2 | Completed | 307 | 27 July 2023 | Kite, A Gilead Company, Santa Monica, CA, USA |
| NCT02445248 | CD19-specific CAR-T cells | Relapsed or refractory DLBCL | Phase 2 | Completed | 115 | 22 December 2022 | Novartis Pharmaceuticals, Basel Switzerland | |
| NCT02601313 | CD19-specific CAR-T cells | Relapsed/refractory mantle cell lymphoma | Phase 2 | Active, not recruiting | 105 | July 2025 | Kite, A Gilead Company | |
| NCT02614066 | CD19-specific CAR-T cells | Relapsed/refractory B-precursor acute lymphoblastic leukemia | Phase 1 Phase 2 | Active, not recruiting | 125 | November 2034 | Kite, A Gilead Company | |
| NCT02631044 | CD19-specific CAR-T cells | Non-Hodgkin lymphoma, diffuse large B cell lymphoma, follicular lymphoma, mantle-cell lymphoma, primary mediastinal B-cell lymphoma | Phase 1 | Active, not recruiting | 385 | 10 May 2024 | Juno Therapeutics, a Subsidiary of Celgene, Seattle, WA, USA | |
| NCT02926833 | CD19-specific CAR-T cells in combination with PD-1 antibodies | DLBCL | Phase 1 Phase 2 | Completed | 37 | 12 January 2023 | Kite, A Gilead Company | |
| NCT03105336 | CD19-specific CAR-T cells | Refractory/relapse large B cell lymphoma | Phase 2 | Active, not recruiting | 159 | Spetember 2036 | Kite, A Gilead Company | |
| NCT03287817 | CD19- and CD22-specific CAR-T cells, followed by anti-PD1 antibody | DLBCL | Phase 1 Phase 2 | Active, not recruiting | 73 | November 2024 | Autolus Limited, London, UK | |
| NCT03310619 | CD19-specific CAR-T cells | Aggressive B-NHL | Phase 1 Phase 2 | Completed | 62 | 15 February 2023 | Celgene, Summit, NJ, USA | |
| NCT03568461 | CD19-specific CAR-T cells | Refractory follicular lymphoma | Phase 2 | Active, not recruiting | 98 | 22 May 2025 | Novartis Pharmaceuticals | |
| NCT00004178 | CEA CAR | Adenocarcinoma | Solid tumor | Phase 1 | Completed | Not given | December 2001 | Roger Williams Medical Center, Providence, RI, USA |
| NCT00019136 | Folate receptor CAR ± IL-2 | Ovarian cancer | Phase 1 | Completed | Not given | Not given | National Cancer Institute (NCI), Rockville, MD, USA | |
| NCT00085930 | GD2 CAR, EBV T cells | Neuroblastoma | Phase 1 | Active, not recruiting | 19 | December 2023 | Baylor College of Medicine, Houston, TX, USA | |
| NCT00730613 | IL-13Ra2 targeting CAR-T cells | Glioblastoma with Hy/TK suicide switch | Phase 1 | Completed | 3 | August 2011 | City of Hope Medical Center, Duarte, CA, USA | |
| NCT00889954 | HER2 CAR, EBV T cells + TGFb DNR | HER2-positive lung cancer | Phase 1 | Completed | 20 | 21 January 2018 | Baylor College of Medicine | |
| NCT00902044 | HER2 CD28 CAR | HER2-positive sarcoma | Phase 1 | Active, not recruiting | 36 | July 2032 | Baylor College of Medicine | |
| NCT01109095 | Her2 CAR, CMV T cells | HER2-positive glioblastoma | Phase 1 | Completed | 16 | 7 March 2018 | Baylor College of Medicine | |
| NCT01140373 | PSMA CAR 2nd | Castrate metastatic | Phase 1 | Active, not recruiting | 13 | June 2024 | Memorial Sloan Kettering Cancer Center, New York, NY, USA | |
| NCT01373047 | CEA CAR | CEA-positive liver metastases | Phase 1 | Completed | 8 | July 2013 | Roger Williams Medical Center | |
| NCT01454596 | EGFRvIII CAR 3rd 28 and 4-1BB ± IL-2 | Glioblastoma | Phase 1 Phase 2 | Completed | 18 | 17 January 2019 | National Cancer Institute (NCI) | |
| NCT01460901 | GD2 CAR multivirus specific | Post-allo HSCT neuroblastoma | Phase 1 | Completed | 5 | January 2015 | Children’s Mercy Hospital Kansas City, Kanzas City, MO, USA | |
| NCT01822652 | GD-2-CAR-T with iCaspase9 Suicide safety switch | Neuroblastoma | Phase 1 | Active, not recruiting | 11 | October 2030 | Baylor College of Medicine | |
| NCT02414269 | Meso-CART cells, modified with iCasp9/M284 | Malignant pleural disease | Phase 1 Phase 2 | Active, not recruiting | 113 | 30 April 2024 | Memorial Sloan Kettering Cancer Center | |
| NCT03170141 | EGFRvIlI-specific CAR-T cells producing PD-1 and PD-L1 antibodies | Glioblastoma multiforme | Phase 1 | Enrolling by invitation | 20 | 31 Decemebr 2023 | Shenzhen Geno-Immune Medical Institute, Shenzhen, China |
References
- Kratzer, T.B.; Jemal, A.; Miller, K.D.; Nash, S.; Wiggins, C.; Redwood, D.; Smith, R.; Siegel, R.L. Cancer statistics for American Indian and Alaska Native individuals, 2022: Including increasing disparities in early onset colorectal cancer. CA Cancer J. Clin. 2023, 73, 120–146.
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365.
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398.
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554.
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56.
- Zhao, J.; Song, Y.; Liu, D. Clinical trials of dual-target CAR T cells, donor-derived CAR T cells, and universal CAR T cells for acute lymphoid leukemia. J. Hematol. Oncol. 2019, 12, 17.
- Mancikova, V.; Smida, M. Current State of CAR T-Cell Therapy in Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2021, 22, 5536.
- Wang, D.; Wang, J.; Hu, G.; Wang, W.; Xiao, Y.; Cai, H.; Jiang, L.; Meng, L.; Yang, Y.; Zhou, X.; et al. A phase 1 study of a novel fully human BCMA-targeting CAR (CT103A) in patients with relapsed/refractory multiple myeloma. Blood 2021, 137, 2890–2901.
- Mei, H.; Li, C.; Jiang, H.; Zhao, X.; Huang, Z.; Jin, D.; Guo, T.; Kou, H.; Liu, L.; Tang, L.; et al. A bispecific CAR-T cell therapy targeting BCMA and CD38 in relapsed or refractory multiple myeloma. J. Hematol. Oncol. 2021, 14, 161.
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354.
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851.
- McLellan, A.D.; Ali Hosseini Rad, S.M. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol. Cell Biol. 2019, 97, 664–674.
- Ajina, A.; Maher, J. Strategies to Address Chimeric Antigen Receptor Tonic Signaling. Mol. Cancer Ther. 2018, 17, 1795–1815.
- James, S.E.; Greenberg, P.D.; Jensen, M.C.; Lin, Y.; Wang, J.; Till, B.G.; Raubitschek, A.A.; Forman, S.J.; Press, O.W. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J. Immunol. 2008, 180, 7028–7038.
- Brentjens, R.J.; Santos, E.; Nikhamin, Y.; Yeh, R.; Matsushita, M.; La Perle, K.; Quintás-Cardama, A.; Larson, S.M.; Sadelain, M. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin. Cancer Res. 2007, 13, 5426–5435.
- Finney, H.M.; Akbar, A.N.; Lawson, A.D. Activation of resting human primary T cells with chimeric receptors: Costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J. Immunol. 2004, 172, 104–113.
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684.
- Teng, M.W.; Kershaw, M.H.; Moeller, M.; Smyth, M.J.; Darcy, P.K. Immunotherapy of cancer using systemically delivered gene-modified human T lymphocytes. Hum. Gene Ther. 2004, 15, 699–708.
- Tang, X.Y.; Sun, Y.; Zhang, A.; Hu, G.L.; Cao, W.; Wang, D.H.; Zhang, B.; Chen, H. Third-generation CD28/4-1BB chimeric antigen receptor T cells for chemotherapy relapsed or refractory acute lymphoblastic leukaemia: A non-randomised, open-label phase I trial protocol. BMJ Open 2016, 6, e013904.
- Zhong, X.S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 2010, 18, 413–420.
- Maus, M.V.; June, C.H. Making Better Chimeric Antigen Receptors for Adoptive T-cell Therapy. Clin. Cancer Res. 2016, 22, 1875–1884.
- Yu, S.; Li, A.; Liu, Q.; Li, T.; Yuan, X.; Han, X.; Wu, K. Chimeric antigen receptor T cells: A novel therapy for solid tumors. J. Hematol. Oncol. 2017, 10, 78.
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154.
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359.
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. eBioMedicine 2020, 58, 102931.
- Ellis, G.I.; Sheppard, N.C.; Riley, J.L. Genetic engineering of T cells for immunotherapy. Nat. Rev. Genet. 2021, 22, 427–447.
- McGuirk, J.; Waller, E.K.; Qayed, M.; Abhyankar, S.; Ericson, S.; Holman, P.; Keir, C.; Myers, G.D. Building blocks for institutional preparation of CTL019 delivery. Cytotherapy 2017, 19, 1015–1024.
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009, 17, 1453–1464.
- Pang, Y.; Hou, X.; Yang, C.; Liu, Y.; Jiang, G. Advances on chimeric antigen receptor-modified T-cell therapy for oncotherapy. Mol. Cancer 2018, 17, 91.
- Riet, T.; Holzinger, A.; Dörrie, J.; Schaft, N.; Schuler, G.; Abken, H. Nonviral RNA transfection to transiently modify T cells with chimeric antigen receptors for adoptive therapy. Methods Mol. Biol. 2013, 969, 187–201.
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028.
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724.
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75.
- Brentjens, R.J.; Latouche, J.B.; Santos, E.; Marti, F.; Gong, M.C.; Lyddane, C.; King, P.D.; Larson, S.; Weiss, M.; Rivière, I.; et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat. Med. 2003, 9, 279–286.
- Lamers, C.H.; Sleijfer, S.; van Steenbergen, S.; van Elzakker, P.; van Krimpen, B.; Groot, C.; Vulto, A.; den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. 2013, 21, 904–912.
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733.
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra138.
- Adusumilli, P.S.; Cherkassky, L.; Villena-Vargas, J.; Colovos, C.; Servais, E.; Plotkin, J.; Jones, D.R.; Sadelain, M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 2014, 6, 261ra151.
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117.
- Fala, L. Yescarta (Axicabtagene Ciloleucel) Second CAR T-cell Therapy Approved for Patients with Certain Types of Large B-Cell Lymphoma. 2018. Available online: https://jons-online.com/browse-by-topic/fda-approvals-news-updates/1829-yescarta-axicabtagene-ciloleucel-second-car-t-cell-therapy-approved-for-patients-with-certain-types-of-large-b-cell-lymphoma (accessed on 20 November 2023).
- Warnings, B. US Food and Drug Administration Approves Bristol Myers Squibb’s and Bluebird bio’s Abecma (Idecabtagene Vicleucel), the First Anti-BCMA CAR T Cell Therapy for Relapsed or Refractory Multiple Myeloma. 2021. Available online: https://www.businesswire.com/news/home/20210326005507/en/U.S.-Food-and-Drug-Administration-Approves-Bristol-Myers-Squibb%E2%80%99s-and-bluebird-bio%E2%80%99s-Abecma-idecabtagene-vicleucel-the-First-Anti-BCMA-CAR-T-Cell-Therapy-for-Relapsed-or-Refractory- (accessed on 20 November 2023).
- Cartron, G.; Fox, C.P.; Liu, F.F.; Kostic, A.; Hasskarl, J.; Li, D.; Bonner, A.; Zhang, Y.; Maloney, D.G.; Kuruvilla, J. Matching-adjusted indirect treatment comparison of chimeric antigen receptor T-cell therapies for third-line or later treatment of relapsed or refractory large B-cell lymphoma: Lisocabtagene maraleucel versus tisagenlecleucel. Exp. Hematol. Oncol. 2022, 11, 17.
- Mian, A.; Hill, B.T. Brexucabtagene autoleucel for the treatment of relapsed/refractory mantle cell lymphoma. Expert Opin. Biol. Ther. 2021, 21, 435–441.
- St-Pierre, F.; Gordon, L.I. Lisocabtagene maraleucel in the treatment of relapsed/refractory large B-cell lymphoma. Future Oncol. 2023, 19, 19–28.
More