Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Douglas Ruden and Version 2 by Jessie Wu.
A key objective in biological aging research is to identify biomarkers capable of predicting the biological age (B-age) of different tissues, as an alternative to relying solely on chronological age (C-age). Biological clock technologies are designed to assess the acceleration of biological age (B-age) in diverse cell types, offering a distinctive opportunity in toxicogenomic research to explore the impact of environmental stressors, social challenges, and unhealthy lifestyles on health impairment. These clocks also play a role in identifying factors that can hinder aging and promote a healthy lifestyle.
- biological clocks
- epigenetic clocks
- toxicology
- toxicogenomics
- DNA methylation
1. Epigenetic Clocks
Accumulating evidence suggests that there are plenty of connections between age and DNAm which led to the development of molecular epigenetic clocks representing biological age [1][2][20,21]. Two of the first molecular epigenetic clocks which are robust predictors of chronological age are Horvath [3][22] and Hannum [4][23] calculators which use DNAm data. Both demonstrate strong age correlations (r = 0.96 for Horvath and r = 0.91 for Hannum) and minimal mean deviations from chronological age (3.6 and 4.9 years, respectively) within their respective validation cohorts [3][4][22,23]. Aging is associated with a decrease in global DNAm along with an increase in local methylation at CpG islands and specific promoters [5][6][7][8][24,25,26,27]. Accelerated epigenetic aging is usually measured in DNA from whole blood and is defined as the difference between the B-age and C-age [9][28]. Accelerated B-age is associated with increased mortality and impaired physical and cognitive function [10][29]. The B-clocks based on DNAm are divided into (i) first-generation (G1) clocks (Hannum and Horvath), that are trained to predict C-age; (ii) second-generation (G2) clocks (PhenoAge, GrimAge), that have improved predictive power over G1 clocks and can predict longevity and the onset of age-related diseases; and (iii) third-generation clocks (G3) (DunedinPoAm), that are designed to estimate the pace of aging [11][18].
Many new B-clocks are being continuously developed till date—therefore in this research researchersticle we present a snapshot of the widely used B-clocks as outlined in Figure 1 and Table 1. The article also discusses how the various environmental factors could cause accelerated epigenetic aging as measured by different epigenetic clocks.
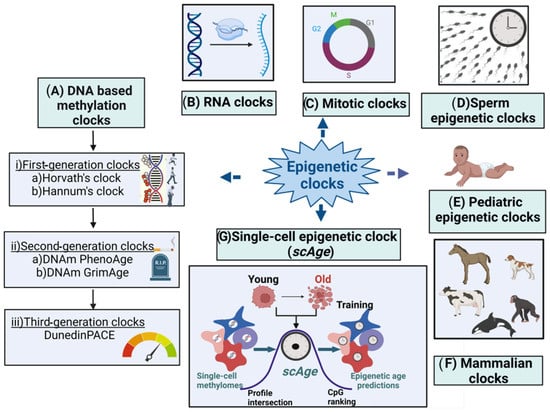
Figure 1. Major types of epigenetic clocks. (A) DNA-based biological clocks: (i) G1 clocks (e.g., Horvath’s and Hannum’s clocks); (ii) G2 clocks (e.g., DNAm PhenoAge and DNAm GrimAge); and (iii) G3 clocks (e.g., DunedinPACE). (B) RNA clocks. (C) Mitotic clocks. (D) Sperm epigenetic clocks. (E) Pediatric epigenetic clocks. (F) Mammalian clocks. (G) Single-cell epigenetic clock (scAge).
Table 1.
An overview of different epigenetic clocks.
| Type of Clock | Name of the Clock |
Publication Year and Ref. |
# of CpG |
No. of Subjects (N) |
Age Range |
Tissue | Training Phenotype |
Platform | |
|---|---|---|---|---|---|---|---|---|---|
| DNAm -based molecular epigenetic clocks |
G1 clocks | Horvath’s clock |
2013; [3][22] | 353 | 7844 | 0–100 | 51 healthy tissues & cell types | C-age | Illumina 27 K and 450 K |
| Hannum’s clock |
2013; [4][23] | 71 | 482 | 19–101 | Whole blood | C-age | 450 K | ||
| G2 clocks | DNAm PhenoAge | 2018; [12][30] | 513 | 9926 | 21–100 | Multiple | Lifespan (mortality risk score) | 27 K and 450 K and EPIC |
|
| DNAm GrimAge | 2019; [13][31] | 1030 | 6935 | 46–78 | Whole blood | Lifespan (mortality risk score) | 450 K and EPIC |
||
| G3 Clocks | Dunedin PACE |
2020; [14][32] | 46 | 810 | 26–38 | Whole Blood | Pace of Aging | 450 K and EPIC |
|
| DNAm- based mitotic clocks |
EpiTOC | 2016; [15][33] | 385 | 656 | 19–101 | Whole blood | Mitotic age, cancer risk | 450 K | |
| MiAge | 2018; [15][33] | 286 | 4020 | N/A | 8 TTGA Cancer cells |
Mitotic age, cancer risk, survival | 450 K | ||
| EpiTOC2 | 2020; [16][34] | 385 | 656 | 19–101 | Whole blood | Mitotic age, cancer risk | 450 K | ||
| Sperm epigenetic clocks |
Sperm epigenetic aging (SEA)CpG clock |
2022; [17][35] | 803,063 | 379 semen samples |
≥18 | Semen | Sperm epigenetic aging | EPIC Infinium Methylation Beadchip |
|
| RNA clocks | RNAAgeCalc | 2020; [18][36] | 353 | 9448 samples | N/A | Multi-tissue | Transcriptional age | RNA-Seq data from the Genotype-Tissue Expression (GTEx) Program | |
| MultiTIMER | 2023; [19][37] | N/A | 23,000 annotated samples | N/A | Multi-tissue | C-age | RNA-seq samples (ArchS4) v11 | ||
| Single-cell epigenetic clock framework (scAge) |
scAge | 2021; [20][38] | 500,000 CpGs/ cell |
549 tissue samples |
1–21 months old mice |
Multi-tissue | Single-cell B-age predictions | Computational platform | |
| Pediatric epigenetic clocks | Knight’s clock | 2016; [21][39] | 148 | 207 | 24–42 weeks | Cord blood |
Gestational Age |
27 K and 450 K | |
| Bohlin’s clock | 2016; [22][40] | 132 | 685 | Neonates | Cord blood | Gestational Age |
450 K | ||
| Lee’s placental clock |
2019; [23][41] | 441,870 | 1102 | 5–42 weeks | Placenta | Gestational Age |
450 K and EPIC | ||
| Pediatric-Buccal-Epigenetic (PedBE) clock | 2020; [24][42] | 94 | 1032 | 0 to 20 years old | Buccal epithelial cels | Pediatric age |
450 K and EPIC | ||
| Mayne’s clock | 2017; [25][43] | 62 | 409 | 8–42 weeks | Placenta | Gestational age |
27 K and 450 K | ||
| NeoAge clocks (4 epigenetic clocks) |
2021; [26][44] | 303–522 | 542 | Pre-term infants (<30 weeks) | Buccal cell samples | Post-menstrual and postnatal age of neonates | 450 K and EPIC | ||
2. First-Generation Molecular Epigenetic Clocks
2.1. DNA Methylation-Based Molecular Epigenetic Clocks
The role of DNAm in the process of aging has been well established [27][28][45,46]. DNAm levels at a subset of loci strongly correlate with age and are applicable to a wide array of tissues, body fluids, and person’s age (from prenatal samples to centenarians) [3][22]. DNAm involves the addition of a methyl group to the 5′ position on cytosines in CpGs [29][47]. The methylation status of millions among the 28 million CpG dinucleotides in the human genome was observed to change as individuals age [30][31][32][33][48,49,50,51]. DNAm-based epigenetic clocks are generally built with a supervised machine learning method and can predict the age of the person or biological sample by combining the methylation values of tens to hundreds of selected CpGs [34][16]. The estimated age, known as B-age or DNAm age, is not only a reflection of C-age but also of the B-age of the DNA source. Biomarkers derived from DNAm provide mechanistic insights into health status, associating the pathogenesis of a disease with biological aging [35][52].
Age acceleration is characterized by variances between age estimates derived from DNAm and chronological age C-age. Positive age acceleration indicates that the DNAm-based age is older than the chronological age, while negative age acceleration signifies a DNAm-based age that is younger than the chronological age. Age acceleration serves as the central focus for all clocks, and reswearchers discuss the most commonly utilized as well as emerging clocks in the following sections.
2.2. Horvath’s Clock
In 2011, a UCLA research team published the first single-tissue DNAm-based age estimator using DNA extracted from saliva [36][53]. The following year, an Italian group discovered using blood a new epigenetic marker of age based on a single CpG [37][54]. In 2013, Horvath and collaborators pioneered the initial multi-tissue predictor of age through the examination of approximately 8000 samples derived from 51 healthy human tissues, including various cell types such as liver, kidney, immune, and brain cells, alongside around 6000 cancer samples. Their findings demonstrated that DNA methylation (DNAm) can serve as a dependable age indicator in normal/non-cancerous tissues [3][22]. Horvath’s DNAm age is calculated based on methylation measurements at 353 loci, present on Illumina’s 450 K and 27 K DNAm microarrays [3][22]. The clock showed remarkable accuracy, predicting age with a correlation of 0.96 and a median error of 3.6 years across various tissues and cells [3][22]. Horvath’s clock stands out for its correlation with C-age across various tissue types, applicability to pediatric samples, strong correlation with gestational age (differentiation day), and the consistency of its age estimates across multiple tissues [38][55].
A recently introduced DNA methylation array, the HorvathMammalMethylChip40, was designed to analyze CpGs that exhibit high conservation across mammals. This array surveys 37,000 CpGs, offering a platform for investigating epigenetic aging in mammals [39][56]. Utilizing this array, researchers have created multi-tissue universal clocks that can be applied to various mammalian species [39][40][56,57]. Using this array, epigenetic clocks for naked mole rats [41][58], elephants [42][59], marsupials [43][60], cats [44][61], pigs [45][62], livestock species (e.g., goat, cattle, sheep, and deer) [46][63], and horses [47][64] were developed. The B-clocks for mice and other mammals were developed using samples from blood and other tissues such as adipose, heart, kidney, liver, lung, spleen, muscle, etc. [48][65]. These readily available B-clocks can be used for wildlife conservation purposes and in breeding programs.
2.3. Hannum’s Clock
In a nearly simultaneous investigation in 2013, Hannum and collaborators constructed a quantitative aging model based on measurements taken from over 450,000 CpG markers in the whole blood of 656 individuals aged 19 to 101 [4][23]. The Hannum clock selectively utilizes 71 CpG sites sourced from the Illumina 450 k array, strategically positioned near genes linked to aging. This targeted selection contributes to its high accuracy in predicting age. The clock has undergone validation in human blood, and ongoing initiatives are focused on adapting the clock for implementation in model organisms like mice [49][66]. Hannum’s age estimator is designed for adult blood samples, resulting in skewed estimates when applied to children and non-blood tissues [4][50][51][23,67,68]. Furthermore, it is affected by age-related changes in blood composition [52][53][5,69].
2.4. Second-Generation Epigenetic Clocks
2.4.1. DNAm PhenoAge
Hannum and colleagues’ blood-based algorithm, along with Horvath and colleagues’ multi-tissue algorithm, generates biological age estimates (DNAm age) that demonstrate a strong correlation with chronological age (C-age), well exceeding r = 0.90 across samples spanning the entire age range. Nevertheless, these biological age estimators display statistically significant associations with numerous age-related health conditions [12][30]. Consequently, Levine et al. developed a DNA methylation-based biomarker referred to as DNAm PhenoAge. This biomarker was designed to predict a surrogate measure of “phenotypic age”, distinguishing between morbidity and mortality risks among individuals of the same chronological age [12][30]. It was devised by calculating a weighted average of 10 clinical characteristics: body mass index (BMI), chronological age (C-age), albumin, creatinine, glucose, and C-reactive protein levels, lymphocyte percentage, cell volume, alkaline phosphatase, and blood cell counts. Subsequently, these values were modeled against DNA methylation (DNAm) levels in blood using a penalized regression model. This approach led to the automatic selection of 513 CpGs, the weighted average of which serves as an effective estimate of phenotypic age. This method represents a significant advancement over first-generation DNA methylation biomarkers (Hannum and Horvath) in predicting morbidity [12][30].
2.4.2. DNAm-Based Biomarker of Mortality (DNAm GrimAge)
In 2019, DNAm GrimAge, an EC based on estimations of plasma protein levels that can predict lifespan and health span was developed by Lu et al. [13][31]. A unique two-stage method was utilized to create DNAm GrimAge. In the initial stage, surrogate DNA methylation biomarkers associated with physiological risk or stress factors were identified. These biomarkers encompass specific plasma proteins: adrenomedullin, C-reactive protein, plasminogen activation inhibitor 1 (PAI-1), and growth differentiation factor 15 (GDF15) [54][55][70,71]. Smoking is a pivotal risk factor for both mortality and morbidity. Consequently, the authors employed a DNA methylation-based estimator of smoking pack-years. In the second stage, these biomarkers were amalgamated into a unified composite biomarker of lifespan known as DNAm GrimAge, expressed in units of years.
A comprehensive meta-analysis, incorporating over 7000 Illumina array measurements, substantiated that DNAm GrimAge surpasses other DNA methylation-based predictors as a more accurate predictor of lifespan. DNAm GrimAge is a linear combination of C-age, sex, and DNA methylation-based surrogate biomarkers for seven plasma proteins and smoking pack-years. It outperforms all other DNA methylation-based biomarkers across a range of health-related metrics [13][31]. The G2 clocks, including DNAm PhenoAge and DNAm GrimAge, surpass current molecular biomarkers of aging due to their robust correlation not only with C-age but also with various age-related conditions [56][15].
2.5. Third-Generation Epigenetic Clocks
A third-generation epigenetic clock, DunedinPACE (Pace of Aging Calculated from the Epigenome), was developed by training on an extensive set of direct biomarkers related to health and diseases. These include BMI, waist-hip ratio, glycated hemoglobin (HbA1c), leptin, blood pressure, lung function, cognitive function, grip strength, and motor coordination, among others. Belsky et al. introduced it in 2021 as an innovative blood biomarker for assessing the pace of aging in the fields of gerontology and geroscience [57][72]. The researchers utilized data from a birth cohort (n = 1037 babies) in Dunedin, New Zealand, born during 1972–1973, tracking them through midlife. Aging was observed to lead to a gradual and progressive deterioration that affected various organ systems. To capture these changes, the authors examined longitudinal shifts in 19 biomarkers, assessing cardiovascular, metabolic, renal, hepatic, immune, dental, and pulmonary systems at ages 26, 32, 38, and 45 years. Analyzing this dataset resulted in the creation of a metric called the “Pace of Aging”, quantifying each participant’s individual rate of biological aging. DunedinPACE demonstrated high test–retest reliability, exhibited associations with morbidity and mortality, and identified accelerated aging in young adults who had experienced childhood adversity [57][72].
DunedinPACE differs from other DNA methylation clocks in both its development methodology and interpretation [57][72]. DunedinPACE contributed additional predictive value beyond clocks like Horvath, Hannum, PhenoAge, or GrimAge in analyzing the occurrence of new cases of morbidity, disability, and mortality. While other clocks estimate aging progression up to a specific point in time, DunedinPACE offers DNAm estimates of the Pace of Aging, representing the ongoing rate of decline in system integrity.
2.6. Mitotic Clocks
The number of cell divisions of a given tissue type in an individual is of great interest since it allows the study of biological aging using a new molecular target (mitotic age) and can predict prospective cancer risk in individuals [58][73]. DNAm-based mitotic-like clocks can keep count of the cumulative number of DNAm errors arising during cell division [15][59][33,74]. There have been three epigenetic mitotic clocks reported in the literature to date: (a) Epigenetic Time to Cancer (EpiTOC), (b) Epigenetic Time to Cancer-2 (EpiTOC2), and (c) Mitotic Age (MiAge) [15][16][58][33,34,73]. In 2016, Yang et al. developed a DNA methylation-based age-correlated model named “EpiTOC” (Epigenetic Timer of Cancer). This model operates as a mitotic clock in both normal and cancerous tissues [15][33]. It utilizes CpGs located in gene promoters associated with the PRC2 polycomb repressive complex, also known as Polycomb group targets (PCGTs), in human embryonic stem cells (hESCs). These CpGs remain constitutively unmethylated in a ground state of age zero across various fetal tissue types. As chronological age (C-age) increases, DNAm levels also rise, enabling researchers to evaluate variations in DNA methylation in both aged and oncogenic tissues.
2.7. Sperm Epigenetic Clocks
Studies have shown that sperm DNAm mediated the age-related effects of male C-age on poor reproductive outcomes like fertilization rates, embryo development, and live birth [60][75]. However, somatic tissue-based clocks cannot precisely predict epigenetic aging in germ cells [3][22]. To address this constraint, Jenkins et al. in 2018 developed the inaugural sperm-specific epigenetic clock within a diverse population, encompassing sperm donors, infertile men, and the community. This was achieved by employing regional methylation levels from the Illumina 450 K array [61][76]. In 2022, Pilsner et al. undertook a more thorough examination, employing the Illumina EPIC array to formulate a novel CpG-based sperm epigenetic age (SEA) within a cohort of 379 participants with the common goal of conception [17][35]. These results indicate that the sperm epigenetic clock could serve as a novel biomarker for the reproductive success of couples. Both the study conducted by Jenkins et al. and that of Pilsner et al. demonstrate that sperm epigenetic age (SEA) is influenced by environmental factors, as evidenced by accelerated SEA observed among smokers in clinical cohorts, sperm donors, and the general public.
2.8. Single-Cell Epigenetic Clock Framework (scAge)
Most existing B-clocks rely on measurements derived from samples containing many cells [62][77]. Bulk samples are valuable for identifying average methylation patterns in tissues, but they neglect the epigenetic heterogeneity that might exist among individual cells [62][63][77,78]. To address this concern, the scAge, an epigenetic clock framework, was devised to allow the profiling of biological age at the level of individual cells [20][38]. Due to the variable coverage of CpGs between cells, the authors employed a ranked intersection algorithm that is agnostic to which CpGs are covered in each cell. This method synthesizes the C-age of tissues while revealing the inherent epigenetic heterogeneity among individual cells. The authors successfully utilized the scAge model to monitor aging in hepatocytes and embryonic fibroblasts, demonstrated reduced epigenetic aging in muscle stem cells, and tracked age dynamics in embryonic stem cells. Consequently, the scAge framework can serve as a valuable tool for exploring epigenetic aging trajectories at single-cell resolution, offering exciting applications in emerging single-cell technologies.
2.9. RNA Clocks
Changes in gene expression in addition to DNAm have also been related with age-related diseases [64][65][66][67][68][69][70][79,80,81,82,83,84,85]. Numerous computer programs exist for predicting DNA methylation (DNAm) age from human DNAm data assessed on the Illumina Infinium HumanMethylation450K BeadChip, such as RNAAgeCalc. However, when it comes to gene expression data, age-related signatures have been developed using either non-human sources or very limited tissue samples [18][36].
2.9.1. RNAAgeCalc: A Multi-Tissue Transcriptional Age Calculator
RNAAgeCalc stands out as the initial versatile transcriptional age calculator that works across tissues and provides tissue-specific predictions. Its development involved utilizing RNA-Seq data from the Genotype-Tissue Expression (GTEx) Program [71][86]. Utilizing publicly available GTEx data, a database that encompasses genes across various tissues, the transcriptional age calculator RNAAgeCalc, was constructed with tissue-specific capabilities [18][36]. The authors subsequently verified, using the calculator, that transcriptional age acceleration was associated with reduced mortality risk and mutation burden in TCGA cancer samples. This provided corresponding insights in comparison to DNAm age [18][36]. Therefore, RNAAgeCalc may advance our efforts for novel treatments for age-related diseases.
2.9.2. Multi-Tissue RNA Clock (MultiTIMER)
It was developed by combining previous data about gene-function associations from the Molecular Biology of the Cell Ontology (MBotC) [72][87] with a machine learning approach to detect the cellular processes and their associated genes which are predictive of age [19][37]. MultiTIMER determines the biological age (B-age) of cells based on their transcriptional profiles, assessing critical cellular processes. Applied to over 70,000 transcriptional profiles, MultiTIMER revealed that the aging process correlates with distinct aging phenotypes and responds to cellular stressors and interventions. In comparison to RNAAgeCalc, which predicts cell age from a set of 1600 genes expressed differentially across tissues during aging, MultiTIMER is more robust [18][36].
2.10. Pediatric Epigenetic Clocks
B-age acceleration measures developed using adult and pediatric samples are not suitable for estimating gestational age (G-age) and are inappropriate for use in neonates. These B-clocks are not trained on G-age data and do not exhibit correlation with G-age. In recent years, various clocks have been specifically designed for predicting G-age and pediatric C-age in pediatric populations [73][88]. Widely used G-age clocks include the Knight clock which was developed from cord blood data using both 27 K and 450 K arrays and consists of 148 CpGs [21][39], and the Bohlin clock which consists of 96 CpGs and was developed using cord blood data from 450 K data [22][40]. The Lee clock was designed using placental data from the 450 K and EPIC arrays and is based on 558 CpGs [23][41]. The Mayne clock was developed using placental data from both 27 K and 450 K arrays and consists of 62 CpGs [25][43]. The NeoAge clock was developed to forecast both postmenstrual age (PMA, time from estimated conception onward) and post-natal age (PNA, time elapsed after birth) for preterm infants. This was achieved using buccal cell samples and considering 303–522 CpGs [26][44]. As far as pediatric clocks are concerned, the Pediatric-Buccal-Epigenetic (PedBE) clock [24][42], comprising 94 CpGs, primarily focuses on the pediatric population (0–20 years old). The PedBE clock was trained in buccal cell samples using the platform of 450 K and EPIC arrays; it is a better predictor when saliva or buccal cells are used compared to blood DNA. The Horvath clock is also an accurate age predictor in children along with adults [3][22].
2.11. Nutrition and Epigenetic Clocks
Exploring nutritional interventions emerges as a promising approach to promote healthy aging, given the growing body of evidence indicating their potential to positively impact the health status of individuals [74][75][89,90]. Key modifiers of epigenetic patterns include dietary factors that supply S-adenosyl-methionine for one-carbon metabolism and polyphenols, such as flavanols, which can inhibit the activity of DNA methyltransferases (DNMTs) [76][91]. Intervention trials were conducted in 44 participants with folic acid + vitamin B12 (GSE74548) and in 13 participants with monomeric and oligomeric flavanols (MOF) (GSE54690) [76][91]. DNAm patterns were evaluated in publicly available Illumina Infinium 450 K methylation datasets. Following supplementation with folic acid + vitamin B12, global DNAm levels were observed to increase in unmethylated regions like CpG islands and shores, while decreasing in highly methylated regions after intervention with MOF. In women with the methylenetetrahydrofolate reductase (MTHFR) 677CC genotype, supplementation with folic acid + vitamin B12 led to a reduction in B-age, as estimated by the Horvath “epigenetic clock” model. Additionally, a population-based prospective cohort study involving 1346 newborns, utilizing Bohlin’s and Knight’s clocks, demonstrated an association between higher maternal plasma homocysteine concentrations and positive G-age acceleration, suggesting a faster epigenetic aging rate compared to clinical gestational aging [77][92]. However, cord serum vitamin B12 concentrations were associated with negative G-age acceleration, indicating slower epigenetic than clinical gestational aging [77][92].
The Mediterranean diet is recognized for its well-balanced combination of nutrients, antioxidants, and anti-inflammatory compounds, and it has been proposed as a potential preventive measure against telomere shortening [78][79][93,94]. A pilot intervention study, involving 120 elderly healthy subjects (60 Italians, 60 Poles), was designed to explore the effects of a Mediterranean-like diet over one year. The diet was specifically tailored to meet the nutritional requirements of individuals over 65 years of age. The study aimed to assess the impact on age-related diseases and functional decline by measuring changes in their biological age (B-age) using Horvath’s clock [80][95]. Polish females and individuals who exhibited higher epigenetic age at baseline appeared to derive greater benefits from the Mediterranean-like diet. Additional research is needed to understand why certain individuals respond more favorably to specific interventions. This is a crucial step in the development of personalized nutritional interventions for precision medicine-based anti-aging strategies.
A 24-month study involving 219 women from the “Diet, Physical Activity, and Mammography” (DAMA) study in Florence, Italy, explored the potential favorable effects of enhanced dietary habits and increased physical activity on the development of breast cancer and aging biomarkers in healthy postmenopausal women aged 50–69 years. [81][96]. DNAm was assessed both at the study’s outset and upon completion. Women engaged in the dietary intervention exhibited a noteworthy deceleration of the DNAm GrimAge clock. Concurrently, increased physical activity resulted in a significant reduction of stochastic epigenetic mutations in critical cancer-related pathways. The DAMA study employed non-extreme interventions, easily achievable through lifestyle modifications. This suggests that policy intervention programs promoting a healthy diet and physical activity could substantially reduce the societal burden of aging-related pathological conditions and diseases. In conclusion, diverse anti-aging strategies, including stress reduction, experimental drugs, and nutritional interventions, can be adopted to decelerate epigenetic aging.