1. Non-Classical Functions of Each Component of the RB-E2F-ARF-MDM2-p53 Pathway
Although the roles of the RB-E2F-ARF-MDM2-p53 pathway in tumor suppression have been well established, accumulating evidence indicates that each component of the pathway has distinct functions, which do not depend on downstream factors. The contribution of these non-classical functions to tumor suppression or promotion has been discerned in mouse models, in which the downstream factor is knocked out. In addition, there are some reports that are contradictory to or challenge the classical views.
2. Non-Classical Functions of RB
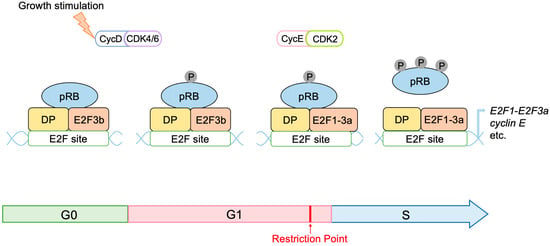
pRB has 14 sites, which are phosphorylated by D-type CDKs. It has been widely accepted that hypo-phosphorylated pRB can bind to and inhibit E2F, whilst hyper-phosphorylation of pRB abolishes binding to E2F. Cyclin D/CDK4, 6 inactivates pRB during the early G1 phase by progressive multi-site phosphorylation of 14 sites to release E2F transcription factors from RB inhibition. However, recent evidence suggests that cyclin D/CDK4, 6 can also activate pRB to bind to and inhibit E2F by phosphorylation at one of the 14 phosphorylation sites (mono-phosphorylation) during the early G1 phase
[1][156] (
Figure 11). Utilizing two-dimensional isoelectric focusing, it has been shown that cells synchronized in the early G1 phase contain exclusively mono-phosphorylated pRB at one of the 14 different phosphorylation sites, catalyzed by cyclin D/CDK4, 6. Moreover, upon DNA damage, mono-phosphorylated pRB, at these distinct phosphorylation sites, that is fully functional in binding to E2F, is observed. At the late G1 restriction point, when the expression of cyclin E and activator E2Fs is induced, cyclin E/Cdk2 inactivates pRB by hyper-phosphorylation, releasing activator E2Fs from pRB binding
[1][156] (
Figure 11). These observations change our understanding of the roles of cyclin D/CDK4, 6 in the regulation of pRB activity with respect to E2F.
Figure 11. Mono-phosphorylation of pRB activates pRB to inhibit E2F. The mono-phosphorylation of pRB by cyclin D/CDK4, 6 activates pRB in binding to and suppressing E2F in the early to mid G1 phase. Additional hyper-phosphorylation by cyclin E/CDK2 inactivates pRB in the binding to and suppression of E2F at the G1/S boundary.
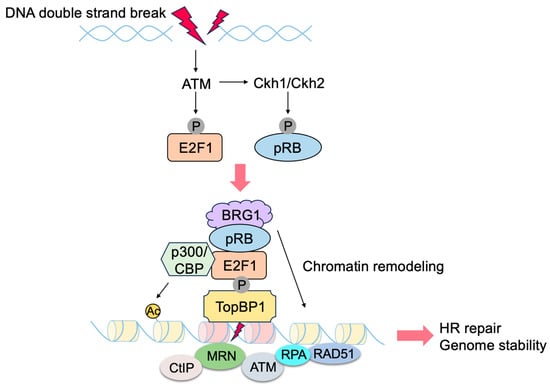
E2F transcriptionally induces the expression of genes involved in checkpoints and DNA repair. In addition, E2F1 and pRB localize to sites of DNA damage and directly promote DNA repair
[2][3][4][157,158,159] (
Figure 12). Upon DNA damage, ATM phosphorylates E2F1 at Ser31
[5][160], and phosphorylated E2F1 binds to a breast cancer susceptibility gene 1 (BRCA1) C terminus (BRCT) domain of DNA topoisomerase 2-binding protein 1 (TopBP1) at the sites of DNA damage
[6][161]. CHK1/CHK2 phosphorylate pRB at Ser612 and facilitate the binding of pRB to E2F1
[7][162], leading to the co-localization of pRB with E2F1 at sites of DNA damage. E2F1 and pRB promote DNA repair by several mechanisms. pRB recruits chromatin remodeling factors such as brahma (BRM)/SWItch 2 (SWI2)-related gene 1 (BRG1), which facilitates DNA end resection and homologous recombination repair of DNA double strand breaks (DSB)
[8][163]. PRB also recruits C-terminal-binding protein (CtBP)-interacting protein (CtIP)
[9][164], which also facilitates DSB end resection
[10][165]. E2F1 and pRB promote the loading of the meiotic recombination 11 (MRE11)-RAD50-Nijmegen breakage syndrome 1 (NBS1) (MRN) complex to sites of DNA double strand breaks and facilitate DNA end resection and the activation of ATM
[11][12][166,167]. E2F1 recruits acetyltransferases p300 and cAMP response element binding protein (CREB)-binding protein (CBP), which facilitates DNA repair by acetylating histones at DNA double-strand breaks
[12][167]. E2F1 co-localizes with RAD51 and replication protein A (RPA) at the sites of DNA DSB and facilitates RAD51-mediated DNA repair
[11][13][166,168]. E2F1 also localizes to sites of UV-induced DNA damage, which is primarily pyrimidine dimers formed in one DNA strand, rather than double strand DNA breaks, and enhances nucleotide excision repair by recruitment of nucleotide excision repair (NER) factors, such as xeroderma pigmentosum, complementation group C (XPC), and XPA
[14][15][169,170]. These results indicate that E2F1, together with pRB, directly promotes DNA repair, contributing to genome stability (
Figure 12).
This review focuses on the role of E2F in linking the RB and p53 pathways; the non-canonical functions of pRB are the subject of other comprehensive reviews
[16][17][18][19][20][21][22][23][24][25][26][27][13,14,171,172,173,174,175,176,177,178,179,180].
Figure 12. pRB and E2F1 directly contribute to genome stability. E2F1, and consequently pRB, are recruited to the sites of DNA damage through TopBP1, which is phosphorylated by ATM upon DNA damage. E2F1 and pRB contribute to DNA repair by recruiting histone acetyltransferase (CBP/p300) and chromatin remodeling factors such as BRG1, respectively.
3. Non-Classical Functions of E2F
3.1. Unique Properties of E2F in Linking the RB Pathway to the p53 Pathway
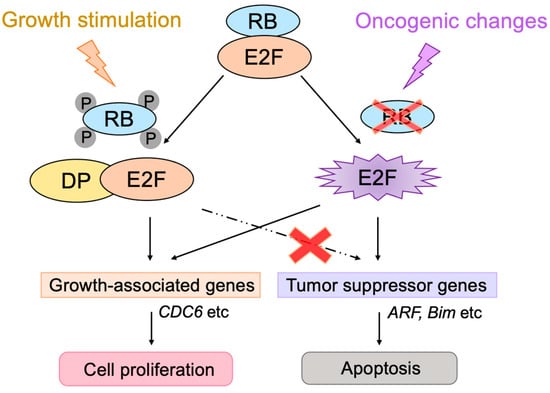
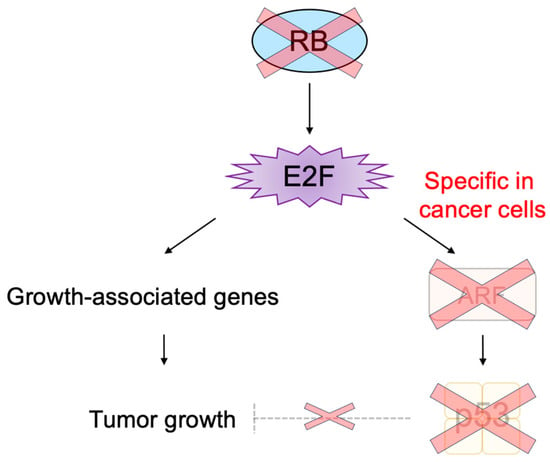
E2F activates a group of growth-related genes upon growth stimulation, thereby playing a central role in cell proliferation. Moreover, upon dysfunction of pRB by oncogenic changes, E2F activates the
Arf tumor suppressor gene to link the RB pathway to the p53 pathway, two major pathways for tumor suppression, thereby playing a crucial role in the suppression of tumorigenesis against oncogenic changes. Intriguingly, serum stimulation, a physiological growth stimulation of fibroblasts, does not activate the
Arf gene, whereas the gene is activated by the over-expression of E2F1 or forced inactivation of pRB by adenovirus E1a or knockdown of pRB by shRNA, which mimic the dysfunction of pRB
[28][181] (
Figure 13). These results indicate that E2F activates the p53 pathway only when the RB pathway is disabled by oncogenic changes to preserve normal cell proliferation in response to physiologic growth signals. Since growth stimulation does not induce deregulated E2F activity, the
Arf gene is not activated by E2F in normal growing cells. Together, deregulated E2F activity, which activates the
Arf gene, is a unique characteristic of cancer cells
[28][181].
The threshold model was suggested to explain the differential regulation of growth-related genes and pro-apoptotic genes by E2F and seems to be widely accepted
[29][10]. It proposes that when the amount of E2F released from RB family members exceeds the first threshold, growth-related genes are activated. Once the amount of free E2F exceeds the second threshold, which is higher than the first one, pro-apoptotic genes are activated in addition to growth-related genes. However, results contradictory to this model have been reported
[30][182]. Activator E2Fs require heterodimeric partner DP to bind to target sequences
[31][38]. According to the threshold model, it is expected that, when the expression of DP is knocked down, the activation of pro-apoptotic genes by over-expressed E2F1 is compromised first, followed by that of growth-related genes. In contrast to this expectation, the knockdown of DP expression significantly reduced E2F1 activation of the growth-related
CDC6 gene but had no effect on the activation of the
Arf gene. These results indicate that the regulation of the
Arf gene, by deregulated E2F, cannot be explained by the amount of free E2F. They also indicate that DP is not necessary for E2F to activate the
Arf gene, building up a distinct activity that activates growth-related genes, which strictly depends on DP
[30][182] (
Figure 13). Hence, deregulated E2F activity, which activates pro-apoptotic genes such as
Arf, is distinct from that which activates growth-related genes.
Figure 13. Distinct regulation of the Arf gene by E2F. Deregulated E2F activity activates the Arf gene, whereas physiological E2F activity induced by growth stimulation does not activate the gene. E2F strictly depends on the heterodimeric partner DP to activate growth-related genes, whereas E2F does not depend on DP to activate the Arf gene. Red cross on RB indicates dysfunction of RB and that on dashed line indicates physiologically induced E2F does not activate tumor suppressor genes.
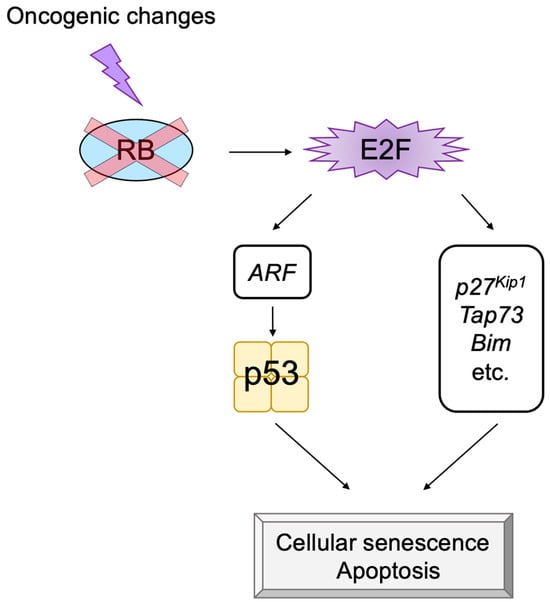
3.2. E2F1 Targets Involved in p53-Independent Pathways for the Induction of Apoptosis
ARF can trigger cellular senescence or apoptosis by activating p53. In addition to the
Arf gene, there are several tumor suppressor genes such as
p27Kip1,
Tap73, and
Bim, which are activated by deregulated E2F activity but not by physiological E2F activity
[32][33][34][183,184,185]. The induction of p27
Kip1 expression by deregulated E2F
[32][183] inhibits CDKs, thereby restraining cell cycle progression
[24][177]. TAp73 is a p53 family member, which can activate p53 target genes independent of p53. The
Tap73 gene is an E2F target
[35][36][186,187] that is specifically activated by deregulated E2F and not by physiological E2F induced by growth stimulation
[33][184]. In addition, there are nine novel targets (
Bim,
Rassf1,
Ppp1r13b,
Jmy,
Moap1,
Rbm38,
Abtb1,
Rbbp4, and
Rbbp7) that are specifically activated by deregulated E2F
[34][185]. Bcl-2 Interacting Mediator of cell death (BIM) is a member of the BH3-only family and destabilizes the mitochondrial outer membrane to release cytochrome
c and trigger apoptosis. Thus, it is expected that deregulated E2F contributes to the induction of cell cycle arrest or apoptosis, not depending on p53, through alternate pathways mediated by p27
Kip1, TAp73, BIM, and other products of the genes listed above, which are specifically induced by deregulated E2F1 (
Figure 14).
Figure 14. Deregulated E2F also activates p53-independent pathways. Deregulated E2F activates not only the Arf gene but also other tumor suppressor genes, which do not depend on p53 to induce cell cycle arrest or apoptosis. Red cross on RB indicates dysfunction of RB.
3.3. Cancer-Cell-Specific Deregulated E2F Activity as a Cancer-Cell-Specific Targeting Tool
The main obstacles in cancer treatment are the side effects caused by current therapies, which also damage normal cells. To avoid these side effects, the researchers have to damage specifically cancer cells and preserve normal growing cells. Almost without exception, the RB pathway is disabled by oncogenic mutations, and E2F activity is enhanced
[37][1]. Enhanced E2F activity promotes a variety of oncogenic processes such as cell proliferation, thereby contributing to tumorigenesis. Therefore, enhanced E2F activity in cancer cells is a fascinating means to specifically target cancer cells
[38][39][188,189]. Enhanced E2F activity in cancer cells has been utilized to express cytotoxic genes (suicide gene therapy) or viral genes essential for the replication of viruses (oncolytic virotherapy) by utilizing growth-related E2F targets, such as the E2F1 promoter
[40][41][42][190,191,192]. However, E2F activity is also enhanced in normal growing cells, since growth stimulation also activates E2F
[43][21]. Thus, this approach may also adversely affect normal growing cells.
E2F activity that activates growth-related genes is enhanced in both cancer cells and normal growing cells. In contrast, E2F activity that activates the
Arf gene specifically exists in cancer cells, since growth stimulation does not activate the tumor suppressor gene in normal growing cells
[28][181] (
Figure 13). Upon dysfunction of pRB by oncogenic changes, deregulated E2F activates the
Arf gene and consequently p53 to induce cellular senescence or apoptosis, protecting cells from tumorigenesis
[44][2] (
Figure 3). In almost all cancers, the ARF-p53 pathway is also disabled, and cancer cells survive
[37][1], leaving deregulated E2F activity tolerated in cancer cells
[28][181] (
Figure 15). Thus, deregulated E2F activity that activates the
Arf gene in cancer cells seems like a more attractive means to specifically target cancer cells than simply enhanced E2F activity that activates growth-related genes. Indeed, it is reported that the ARF promoter, which is not activated by growth stimulation, exhibits lower activity than the E2F1 promoter, which is activated by growth stimulation, in normal growing cells. However, the ARF promoter showed comparable activity to the E2F1 promoter in cancer cells, thereby showing higher cancer cell specificity than the E2F1 promoter
[45][193]. Similarly, recombinant adenovirus expressing the cytotoxic
HSV-TK gene under the control of the ARF promoter exhibited lower cytotoxicity than that with the E2F1 promoter in normal growing cells. However, both constructs showed similar cytotoxicity in cancer cell lines
[45][193]. These observations suggest that utilizing deregulated E2F activity by the ARF promoter is superior to that by the E2F1 promoter to drive gene expression specifically in cancer cells
[46][194].
Figure 15. Deregulated E2F activity, which activates tumor suppressor genes, specifically exists in cancer cells. Upon dysfunction of the RB pathway, deregulated E2F1 activates the Arf gene to protect cells from tumorigenesis. However, the p53 pathway is also disabled in cancer cells, enabling cancer cells to survive, leaving deregulated E2F1 activity that specifically exists in cancer cells. Red crosses on RB, ARF and p53 indicate their dysfunction, and that on dashed line indicates p53 cannot suppress tumor growth.
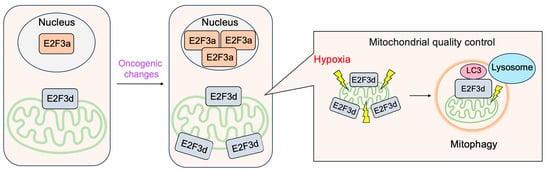
3.4. E2F3d, a Novel Member of the E2F3 Family, Mediates Hypoxia-Induced Mitophagy in Cancer Cells
In addition to E2F3a and E2F3b, novel members of the E2F3 family, E2F3c and E2F3d have been identified
[47][41]. Both isoforms are generated by alternative splicing of E2F3a mRNA, and thus their expression parallels that of E2F3a. Intriguingly, both isoforms lack NLS and DBD and localize in the cytoplasm. E2F3d localizes to the outer membrane of the mitochondria and has an LC3-interacting region (LIR) motif created by a shift in the reading frame due to alternative splicing in its cytosolic domain. These observations suggest the involvement of E2F3d in mitochondrial autophagy (mitophagy). Indeed, the over-expression of E2F3d induces mitochondrial fragmentation and mitophagy. Conversely, the knockdown of E2F3s suppresses mitophagy induced by hypoxia and increases intracellular levels of reactive oxygen species in cancer cells. These results indicate that E2F3d facilitates mitophagy upon hypoxia in cancer cells
[47][41] (
Figure 16).
This review focuses on the roles of E2F in linking the RB and p53 pathways; other comprehensive reviews have addressed the non-canonical functions of E2F
[17][23][29][48][49][50][51][52][53][54][10,12,14,15,16,17,18,19,176,195].
Figure 16. E2F3d facilitates mitophagy upon hypoxia in cancer cells. E2F3d does not possess DBD due to alternative splicing and localizes at the outer membrane of the mitochondria. Due to the frame shift caused by alternative splicing, E2F3d possesses an LC3-interacting region (LIR) motif at the cytosolic domain and functions as a mitophagy receptor upon hypoxia in cancer cells.
4. p53-Independent Functions of ARF in Tumor Suppression
Although p53 is a critical element in ARF-mediated tumor suppression, many lines of evidence indicate that ARF has other important tumor suppressor functions, independent of p53
[55][56][196,197].
Arf,
Tp53, and
Mdm2 triple knockout mice show a higher frequency of tumor formation than
Tp53 and
Mdm2 double knockout mice, demonstrating that ARF can function independently of MDM2 and p53 in tumor suppression
[57][198]. P14
ARF inhibits the proliferation of p53-null human cancer cell lines by inducing G2/M arrest and subsequently apoptosis in tissue cultures and in mouse xenograft models
[58][199]. The introduction of p14
ARF into human
Arf-silenced U-2 OS and p53(−/−) Saos-2 cells induces apoptosis in response to IFN-β treatment, which is not inhibited by the expression of dominant negative p53, suggesting that the induction of apoptosis by IFN-β requires an ARF function that is independent of p53
[59][200]. In addition, in a mouse xenograft model of pancreatic ductal adenocarcinoma metastasis, ARF inhibits tumor cell colonization independently of p53
[60][201]. These observations suggest that there are targets other than the MDM2-p53 axis for tumor suppression mediated by ARF.
4.1. ARF Suppresses Ribosomal Biogenesis
ARF suppresses ribosomal biogenesis, thereby reducing cell proliferation (
Figure 17). P19
ARF inhibits the production of ribosomal RNA (rRNA) and processing, independent of p53, thereby inhibiting cell proliferation
[61][202]. The reduction of rRNA synthesis may be due, at least in part, to the binding of ARF to the rRNA gene promoter along with topoisomerase I, which functions in rRNA transcription, as shown by the chromatin immunoprecipitation assay
[62][203]. In addition, ARF inhibits the import of the RNA polymerase I transcription termination factor (TTF-I) into the nucleolus by binding to the nucleolar localization sequence (NoLS)
[63][204]. ARF also suppresses ribosomal biogenesis through interaction with nucleophosmin (NPM), which is thought to facilitate the maturation of pre-ribosomal particles and the nuclear export of ribosomes
[64][65][66][67][205,206,207,208]. The association of ARF and NPM occurs in the absence of MDM2 and p53 and is antagonized by MDM2. The knockdown of NPM reduces the amount of ARF in the nucleolus and facilitates ARF-MDM2 interaction in the nucleoplasm, which correlates with growth suppression and p53 activation mediated by ARF. Conversely, over-expression of NPM increases nucleolar ARF and inhibits ARF function. These observations suggest that NPM targets ARF to nucleoli and inhibits its function
[68][209]. Thus, NPM and ARF antagonize one another to either promote or inhibit cell growth. Indeed, in
Arf(−/−) cells, ribosome biogenesis and protein synthesis are enhanced, leading to increased cell protein and volume, which are reversed by the knockdown of NPM
[69][210]. ARF also binds to DDX5, which was originally identified as an RNA helicase and also functions as a transcriptional coactivator. ARF reduces the amount of nucleolar DDX5, which is required for transcription and maturation of rRNA, and also inhibits the association of DDX5 with the rDNA promoter and nuclear pre-ribosomes. Thus, the inhibition of DDX5 by ARF results in reduced ribosome genesis and cell proliferation
[70][211]. Hepatocyte odd protein shuttling (HOPS), whose over-expression results in cell cycle arrest in G0/G1 and whose knockdown causes centrosome hyper-amplification, interacts with NPM and p19
ARF, functioning as a bridging protein between NPM and p19
ARF, which are mutually antagonistic with respect to tumor cell proliferation
[71][212]. ARF also suppresses the expression of Drosha, a known DDX5 interacting protein, and attenuates the maturation of miRNAs and ribosomal RNAs (rRNAs), by suppressing the translation of Drosha mRNAs
[72][213]. ARF-null cells, transformed by oncogenic Ras(V12), show an increased expression of Drosha, and the knockdown of Drosha inhibits Ras-dependent cellular transformation
[73][214]. These observations point to the critical roles of ARF in the suppression of cell growth by inhibiting ribosome biogenesis.
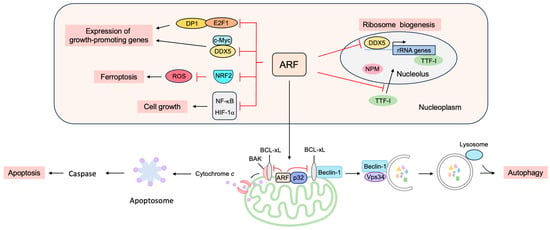
Figure 17. p53-independent functions of ARF in tumor suppression. ARF can suppress cell proliferation independently of p53, such as by the suppression of ribosome biogenesis and growth-promoting transcription factors and the induction of apoptosis, autophagy, and ferroptosis.
4.2. ARF Suppresses the Expression of Growth-Promoting Genes
ARF suppresses gene expression by binding to several transcription factors and coactivators to suppress both the transcription and translation of mRNAs (Figure 17).
ARF suppresses E2F, the central player in cell cycle progression
[74][78]. ARF can inhibit E2F activity in p53(−/−) cells, indicating that the suppression of E2F activity is not dependent on p53. P14
ARF binds to E2F1 and suppresses its transcriptional activity
[75][76][77][215,216,217], while p19
ARF binds directly to DP1, a heterodimeric partner of E2F, and inhibits the interaction between DP1 and E2F1
[78][218]. Since E2F1 requires DP1 for high affinity binding to growth-related target genes, this results in a reduction in E2F target gene expression and cell cycle arrest. Thus, DP1 also seems to be a critical direct target of ARF
[79][219]. In addition, p19
ARF binds to E2F1–E2F3 and facilitates degradation through the proteasome, thereby reducing E2F transcriptional activity
[80][220]. Taken together, the binding to and suppression of E2F seems to be an important function of ARF, independent of p53, for growth suppression.
ARF interacts with c-Myc independently of MDM2 or p53 and translocates c-Myc from the nucleoplasm to the nucleolus, thereby inhibiting its transcriptional activity
[81][221]. ARF binding to DDX5 also inhibits its coactivator function. DDX5 binds to c-Myc and enhances its transcriptional activity. ARF binds to DDX5 and blocks its association with c-Myc, reducing c-Myc-mediated gene expression
[82][222]. P14
ARF inhibits the transcriptional activity of hypoxia-inducible factor (HIF)-1α, which plays a crucial role in the adaptation of tumor cells to hypoxia by sequestering it into the nucleolus
[83][223]. ARF also inhibits the transcriptional activity of nuclear factor-kappa B (NF-κB), which plays a critical role in cell survival, independent of MDM2 and p53. ARF inhibits the transcriptional activity of the NF-κB family member RelA by recruiting histone deacetylase 1 (HDAC1)
[84][224]. ARF activates ATR and CHK1, which phosphorylate the transactivation domain of RelA at threonine 505, and it suppresses transcriptional activity
[85][225]. ARF also binds to and suppresses protein phosphatase 1G (PPM1G), which functions as a coactivator of NF-κB
[86][226]. In addition, p19
ARF suppresses vascular endothelial growth factor A (VEGFA) expression through repression of the translation of VEGFA mRNA, thereby suppressing tumor angiogenesis. Translational repression of VEGFA mRNA by p19
ARF depends on binding to NPM but does not require p53
[87][227]. P19
ARF binds to the anti-apoptotic transcriptional corepressor C-terminal binding protein (CtBP), leading to proteasome-dependent degradation of CtBP. Consistent with this, ARF expression and knockdown of CtBP by siRNA led to p53-independent apoptosis in colon cancer cells
[88][228]. The pro-apoptotic genes
Bik,
Bim, and
Bmf are repressed by CtBP, which is reversed by ARF
[89][229]. P14
ARF binds to the androgen receptor (AR) and suppresses transactivation in prostate cancer cells
[90][230]. P14
ARF interacts with the Myc-interacting zinc finger protein 1 (MIZ1) and facilitates the formation of a heterochromatic complex containing MIZ1, c-Myc, and trimethylated H3K9, leading to the repression of the genes involved in cell adhesion and signal transduction as well as the induction of apoptosis
[91][231].
Reactive oxygen species (ROS) levels are tightly controlled by the transcription factor nuclear factor E2-related factor 2 (NRF2). In response to increased ROS levels, NRF2 activates target genes involved in the inactivation of ROS. The activation of oncogenes such as K-Ras(G12D), B-Raf(V619E) and Myc(ERT2) increases NRF2 activity to engage and enhance the antioxidant program, thereby lowering intracellular ROS
[92][232]. ARF binds to NRF2 and inhibits the expression of its target genes, including
Slc7a11. SLC7A11 is a key component of the cystine/glutamate antiporter, and cystine uptake is critical for glutathione synthesis to inactivate ROS. The suppression of SLC7A11 expression reduces intracellular cysteine levels and renders cells unable to cope with oxidative stress and susceptible to ferroptosis. Thus, ARF sensitizes cells to ferroptosis independent of p53
[93][233].
4.3. ARF Facilitates Apoptosis at the Mitochondria
Although ARF is primarily located in the nucleolus, ARF translocates to the nucleoplasm and into the cytoplasm in response to a variety of stresses. In the cytoplasm, ARF localizes to the mitochondria (
Figure 17). The mitochondrial protein p32 binds to the C-terminal region of ARF, encoded by exon 2, and recruits ARF to the mitochondria. The localization of ARF to the mitochondria reduces the mitochondrial outer membrane potential and induces apoptosis. Mutations in exon 2 of
Arf, which are frequently observed in human cancers, compromise the localization of ARF to the mitochondria and the consequent induction of apoptosis. These observations may explain the frequent mutations of exon 2 in human cancers
[94][234]. Interestingly, the interaction of ARF with p32 is not required for the mitochondrial accumulation of ARF, and highly hydrophobic domains within the amino-terminal half of p14
ARF serve as mitochondrial localization sequences. This allows ARF to interact with p32 and Bcl-xL to induce apoptosis (
Figure 17)
[95][235]. ARF also activates BAK by reducing the expression levels of MCL-1 and BCLxL
[96][236]. The C-terminal nuclear/nucleolar localization sequence (NLS/NoLS) of p14
ARF is crucial for nucleolar localization of p14
ARF in the absence of cellular stress. Protein arginine methyltransferase 1 (PRMT1) methylates several arginine residues in the NLS/NoLS of p14
ARF. Genotoxic stress facilitates the interaction of PRMT1 and p14
ARF, leading to the arginine methylation of p14
ARF. The methylation of the NLS/NoLS promotes the release of p14
ARF from the nucleolus, subsequently leading to the interaction with mitochondrial p32 and induction of apoptosis independent of p53
[97][237].
4.4. ARF Facilitates Autophagy at the Mitochondria
Autophagy is a self-digesting process of cells, which degrades unnecessary or dysfunctional components through the lysosomal pathway, enabling energy supply and organelle renewal in response to insufficient nutrients supply
[98][238]. Enforced autophagy often causes cell death (autophagic cell death) and contributes to tumor suppression.
ARF can induce autophagy in cells lacking p53 function
[99][239]. Mitochondrial ARF interacts with the BCL-2 family member BCL-xL, relieving the inhibition of the Beclin-1/Vps34 complex, which is essential for autophagy (
Figure 17)
[100][240]. ARF interacts with cytosolic 70 kDa heat shock protein (HSP70), which mediates the trafficking of ARF to the mitochondria and autophagy
[101][241]. For MDM2-mediated activation of p53, the region of ARF coded by exon 1 is crucial. In contrast, for the induction of autophagy, similar to apoptosis, ARF exon 2 is important, which contains the majority of
Arf mutations in human cancer
[102][242], suggesting that the induction of autophagy by ARF also plays an important role in tumor suppression. Autophagy contributes to the survival of cells by adaptation to energy starvation. Consequently, ARF can also contribute to tumor promotion according to cellular circumstances by facilitating autophagy in a p53-independent manner. It is suggested that the phosphorylation of ARF at Thr8 by protein kinase C may control whether ARF promotes or counteracts autophagy, since over-expression of a phosphomimetic mutant ARF(T8D) suppresses autophagy
[103][243].
In addition, a short mitochondrial form of p19
ARF has been identified that induces autophagy and caspase-independent cell death. The short isoform (smARF, short mitochondrial ARF) is produced by translation from an internal initiation codon, Met45, and is devoid of the nucleolar functional domains. The human
p14ARF mRNA also produces the shorter isoform. In response to oncogenic stress, the expression of smARF increases concomitantly with full-length ARF and reduces mitochondrial outer membrane potential without causing cytochrome
c release or caspase activation, leading to the induction of autophagy and cell death
[104][244]. SmARF also interacts with the mitochondrial p32 protein to stabilize the protein
[105][245]. Full-length ARF can cause cellular autophagy, whereas smARF is suggested to selectively induce autophagy of the mitochondria (mitophagy)
[102][242].
4.5. ARF Contributes to Genome Stability
ARF seems to contribute to the maintenance of chromosomal stability. The nuclear interactor of ARF and MDM2 (NIAM) binds both ARF and MDM2. NIAM activates p53 in collaboration with ARF and causes a G1 phase cell cycle arrest. NIAM also inhibits cell growth in cells lacking p53, and knockdown experiments shows that NIAM is not essential for ARF-mediated growth inhibition. These results suggest that NIAM and ARF act both in the same and distinct pathways to cooperatively suppress cell growth. Intriguingly, the knockdown of NIAM accelerates chromosomal instability, indicating a role in maintaining chromosomal stability
[106][246]. ARF also plays p53-independent roles in the mitotic checkpoint to maintain chromosomal stability. Aneuploidy is induced upon the loss of ARF function both in vitro and in vivo. ARF-null mouse embryonic fibroblasts (MEFs) show mitotic defects such as misaligned and lagging chromosomes, multipolar spindles, and tetraploidy, likely due to the over-expression of Aurora B. The mitotic defects of ARF-null cells can be restored by returning the expression of Aurora B to near-normal levels. These observations indicate that ARF plays an important role in chromosome segregation and the mitotic checkpoint by restraining Aurora B, thereby maintaining chromosomal stability
[107][247].
4.6. ARF Facilitates the SUMOylation of Interacting Proteins
Much of the p53-independent functions of ARF appear to be mediated by the SUMOylation of its interacting proteins. ARF induces the SUMOylation of interacting proteins, including MDM2 and NPM, independently of p53
[108][116]. The introduction of ARF in
Arf(−/−) NIH 3T3 cells induces the SUMOylation of MDM2 and NPM before p53-dependent cell cycle arrest. The binding of ARF to MDM2 and NPM and the nucleolar localization of ARF is required for ARF to SUMOylate MDM2 and NPM. The inhibition of the SUMO activating enzyme (E1) or knockdown of the SUMO conjugating enzyme (E2/Ubc9) blocked ARF-induced SUMOylation of MDM2, but had no effect on ARF activation of p53. These observations suggest that the p53-independent effects of ARF on gene expression and tumor suppression might depend on the ARF-induced SUMOylation of interacting proteins
[109][248].
P63 is a member of the p53 family, whose gene encodes two isoforms; one has a transcriptional activation domain in the N-terminal region (TAp63), and the other lacks this domain but has another transcriptional domain at the extreme N-terminus (ΔNp63). TAp63 functions in the development of tissues, such as skin, by activating target genes. In contrast, ΔNp63 suppresses not only TAp63 but also p53, thereby promoting cell growth and contributing to tumorigenesis. p14
ARF interacts with both TAp63 and ΔNp63 through their N-terminal trans-activation domains and inhibits p63-mediated trans-activation and trans-repression
[110][249]. p14
ARF also destabilizes certain p63 isoforms. ΔNp63α is the most abundantly expressed p63 isoform, and p14
ARF targets ΔNp63α to proteasomal degradation by stimulating its SUMOylation. ΔNp63 is preferentially SUMOylated by SUMO2, and p14
ARF increases the efficiency of this process
[111][250]. For efficient proteasomal degradation of ΔNp63α, both SUMOylation and ubiquitylation are required
[112][251].
ARF stimulates the SUMOylation of NPM, which facilitates ribosome biogenesis, thereby suppressing ribosome biogenesis. The nucleolar SUMO-specific protease SUMO1/sentrin/SMT3 specific peptidase 3 (SENP3) associates with NPM and catalyzes the deSUMOylation of NPM-SUMO conjugates. The knockdown of SENP3 interferes with nucleolar ribosomal RNA processing and inhibits the conversion of the 32S rRNA species to the 28S form, which is also observed on the depletion of NPM. These observations suggest that SENP3 plays critical roles in ribosome biogenesis by deconjugating SUMO from NPM
[113][252]. Dependent upon the presence of NPM, p19
ARF triggers sequential phosphorylation, polyubiquitination, and rapid proteasomal degradation of SENP3. Thus, p19
ARF and SENP3 act in opposition to each other in the SUMOylation of target proteins such as NPM
[114][253].
Taken together, ARF fulfills a variety of MDM2-p53 independent functions in the suppression of cell proliferation (Figure 17). It is expected that the induction of Arf gene expression by deregulated E2F, upon oncogenic changes, not only activates p53, through the inactivation of MDM2, but also activates MDM2-p53-independent functions of ARF to suppress tumorigenesis.
5. P53-Independent Functions of MDM2 in Tumor Promotion
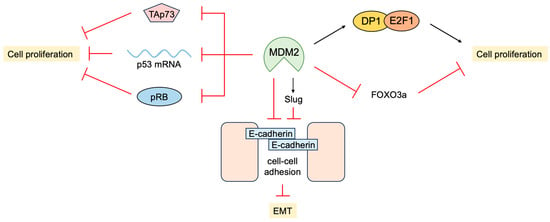
In addition to the inhibition of transcriptional activity and facilitating the degradation of p53, MDM2 promotes cell proliferation independent of p53. MDM2 physically interacts with pRB and inhibits its ability to suppress cell proliferation
[115][254]. Interestingly, pRB-bound MDM2 can still bind to p53 and suppress transcriptional activation, but this does not affect transcriptional repression and promotes p53-mediated apoptosis
[116][255]. MDM2 also binds to E2F1 and DP1 through the activation domain of E2F1. In contrast to binding to p53, MDM2 binding to E2F1/DP1 stimulates its transcriptional activity
[117][256]. Interestingly, MDM2 suppresses apoptosis induced by E2F1/DP1 but stimulates DNA synthesis and colony formation depending on DP1
[118][257]. These results indicate that MDM2 not only inhibits growth suppression by p53 but also promotes cell proliferation by enhancing E2F1/DP1 activity. Consistent with this, the over-expression of MDM2 in the mammary gland of mice disturbs its normal development and morphogenesis regardless of the presence of p53. This may be due to the over-expression of MDM2 promoting the S phase in the absence of mitosis, thereby generating polyploid cells
[119][258].
MDM2 binds to not only the p53 protein but also p53 mRNA. This interaction is through the RING domain of MDM2 and impairs its E3 ligase activity but promotes the translation of p53 mRNA
[120][259]. ATM phosphorylates MDM2 at Ser395 and facilitates the MDM2-p53 mRNA interaction. The Ser395 phosphorylation of MDM2 also promotes its SUMOylation and nucleolar accumulation, which facilitates p53 stabilization and activation following DNA damage
[121][260]. MDM2 can also inhibit the translation of p53 mRNA by regulating ribosomal protein L26, which binds to p53 mRNA and facilitates its translation
[122][261]. MDM2 binds to and polyubiquitylates L26, facilitating its degradation through proteasome. MDM2 also inhibits the interaction of L26 with p53 mRNA, thereby suppressing p53 protein production. Genotoxic stress suppresses the inhibitory effects of MDM2 on L26, thereby rapidly increasing p53 protein production
[123][262].
pRB and E2F1 bind the MRE11-RAD50-NBS1 (MRN) complex through NBS1 and facilitate DNA double-strand break repair
[4][159]. MDM2 also binds to NBS1, independent of ARF or p53, and co-localizes with NBS1 to the sites of DNA damage. However, the over-expression of MDM2 delays DNA double-strand break repair, suggesting that MDM2 is negatively involved in the repair process and genomic stability
[124][263].
MDM2 promotes epithelial-mesenchymal transition (EMT) and invasiveness by several mechanisms. MDM2 binds directly to E-cadherin and down regulates its expression by ubiquitination and endosomal degradation. The over-expression of MDM2 in breast cancer cells disrupts cell-to-cell contacts and increases cell motility and invasiveness
[125][264]. MDM2 mediates the downregulation of Forkhead box O 3a (FOXO3a) expression by the RAS-extracellular signal-regulated kinase (ERK) pathway through ubiquitin-proteasome-mediated degradation of ERK-phosphorylated FOXO3a, leading to enhanced cell proliferation and tumorigenesis
[126][265]. In addition, MDM2 enhances the expression of the transcription factor Slug, which suppresses the expression of E-cadherin and promotes EMT by binding to and stabilizing the Slug mRNA
[127][266]. In contrast, p53 induces the expression of MDM2, which binds to Slug along with p53, and facilitates MDM2-mediated degradation of Slug, thereby suppressing cancer cell invasion
[128][267].
MDM2 and MDM4 also bind to TAp73 and suppress its transcriptional activity; however, MDM2 and MDM4 do not facilitate the degradation of TAp73 but rather stabilize the protein
[129][130][131][132][268,269,270,271]. Consistent with this, the inhibition of MDM2 or MDM4 in cells lacking p53 function causes cell cycle arrest, despite reduced levels of TAp73
[133][272], likely due to release from transcriptional repression. In addition, the inactivation of MDM2 in p53-deficient triple-negative breast cancer cells induces apoptosis mediated by TAp73
[134][273]. These observations suggest that MDM2 and MDM4 promote cell proliferation, at least in part, through the inhibition of TAp73.
Taken together, MDM2 and MDM4 have a variety of p53-independent functions, which may contribute to cell proliferation and tumorigenesis (Figure 18). It is reasonable to predict that the induction of ARF inhibits these functions of MDM2 and MDM4, in addition to the suppression of p53.
Figure 18. p53-independent functions of MDM2 in tumor promotion. MDM2 binds to and suppresses growth-inhibiting factors such as pRB, TAp73, and p53 mRNA. In addition, MDM2 also binds to and enhances growth-promoting factors such as E2F1/DP1. Moreover, MDM2 promotes epithelial-mesenchymal transition (EMT) by the downregulation of FOXO3a and induction of Slug expression.
6. Non-Classical Functions of p53
6.1. Novel p53 Targets Genes Important for Tumor Suppression
It is generally accepted that p53 plays a crucial role in tumor suppression by inducing cell cycle arrest or apoptosis, mediated by the transcriptional activation of the
p21Cip1 and
Puma or
Noxa genes, respectively. The requirement for these three genes was examined using triple knockout mice (p21−/−puma−/−noxa−/− mice)
[135][274]. As expected, p53 could not induce cell cycle arrest, cellular senescence, or apoptosis in the cells obtained from these mice. However, these mice did not develop tumors up to 500 days, whereas p53-null mice all developed tumors by 250 days. This observation suggests that there are other important p53 targets for tumor suppression. Interestingly, DNA damage induced by γ irradiation remained longer in p53-null cells than in wild-type or triple knockout (p21−/−puma−/−noxa−/−) cells. In addition, p53-null cells could not induce expression of several p53 target genes involved in DNA repair. These observations indicate that the induction of cell cycle arrest, cellular senescence, or apoptosis is not sufficient for tumor suppression mediated by p53 in vivo and that maintaining genomic stability and likely other processes may be important
[135][136][274,275].
The ShRNA-mediated knockdown of p53 target genes in vivo in tumor-prone genetic backgrounds identified several DNA repair genes. The combined knockdown of DNA repair genes (
Mlh1,
Msh2,
Rnf144b,
Cav1, and
Ddit4) facilitated tumor formation to a similar extent as the knockdown of p53. Moreover, the knockdown of Mlh1 alone facilitated tumor formation in a wild-type background, and the over-expression of Mlh1 delayed tumor formation caused by the loss of p53. These observations indicate that DNA repair is crucial for p53-mediated tumor suppression
[137][276] (
Figure 19).
In a murine model of pancreatic ductal adenocarcinoma, the
tyrosine-protein phosphatase non-receptor type 14 (
Ptpn14) gene was identified as a p53 target gene, which plays a crucial role in p53-mediated tumor suppression. Ptpn14 suppresses functions of the Yap oncoprotein in the Hippo pathway and is necessary for the suppression of pancreatic carcinogenesis mediated by p53
[138][277].
The cholesterol transport protein ATP-binding cassette, subfamily A, member 1 (ABCA1) mediates the transport of cholesterol from the plasma membrane to the endoplasmic reticulum (ER) and represses sterol regulatory element-binding protein 2 (SREBP-2), the master regulator of the mevalonate pathway. SREBP-2 transcriptionally induces the expression of hydroxymethylglutaryl (HMG)-CoA reductase, the rate limiting enzyme of that pathway. SREBP-2 is anchored in the membranes of the ER as a precursor protein and, upon a reduction in cholesterol levels, is cleaved and released as a mature form to transactivate target genes. The mevalonate pathway produces not only cholesterol but also isoprenoids, which are required for the prenylation of growth signal transmitters to localize to the cell membrane as intermediate metabolites. P53 transactivates the
ABCA1 gene and represses SREBP-2, thereby suppressing the mevalonate pathway
[139][278] (
Figure 19). The knockdown of mevalonate pathway genes, such as
HMG-CoA reductase, and inhibition of HMG-CoA reductase by Atorvastatin suppresses murine hepatocellular carcinogenesis driven by the loss of p53. Similar to p53 loss, CRISPR/Cas9-mediated inactivation of the
ABCA1 gene in mice promotes liver tumorigenesis with enhanced SREBP-2 activation. These observations indicate that the
ABCA1 gene is an important p53 target in the suppression of liver tumorigenesis
[139][278].
The transactivation domain 1 (TAD1) mutant of p53 (p53
25,26) cannot activate classical p53 target genes such as
p21Cip1,
Noxa, and
Puma, but it is able to activate a small subset of targets. Surprisingly, this mutant is fully capable of suppressing tumorigenesis in a knockin mouse model. Thus, the activation of most of the target genes is not necessary for p53 to suppress tumorigenesis
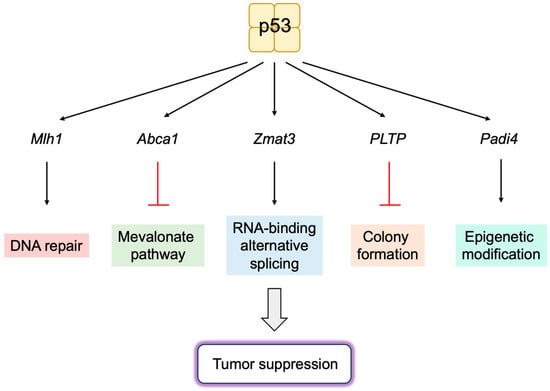
[140][141][279,280]. Utilizing p53
25,26, a novel p53-inducible gene
Zmat3 was identified by RNA interference and CRISPR/Cas9 screening. Zmat3 is an RNA-binding protein, which regulates exon inclusion in transcripts encoding proteins of various functions
[142][281]. The most alternatively spliced mRNA by Zmat3 in colorectal cancer (CRC) cells was CD44, a cell attachment factor and stem cell marker that is involved in tumorigenesis. The inhibition of Zmat3 enhanced the inclusion of CD44 variant exons, generating oncogenic CD44 isoforms (CD44v) and enhanced CRC cell proliferation. The inhibition of p53 also increased CD44v, suggesting that the regulation of CD44 splicing by Zmat3 is critical for p53-mediated tumor suppression
[143][282]. These observations indicate that Zmat3 is a novel RNA-splicing regulator and a crucial target gene of p53 in tumor suppression (
Figure 19).
Studies utilizing hypomorphic mutants of p53, P47S, Y107H, and G334R, which cannot activate a subset of target genes, identified
Phospholipid transfer protein (
Pltp) as an important p53 target gene in growth suppression. The over-expression of PLTP inhibits colony formation of human cancer cell lines. Both p53 and PLTP decrease the sensitivity of cells to ferroptosis in human hepatocellular carcinoma cell line HepG2. These observations suggest that
Pltp is a p53 target gene, which has a role in growth suppression and sensitivity to ferroptosis
[144][283] (
Figure 19).
Similarly, utilizing the variant of p53, Y107H,
Padi4 was identified as an important p53 target gene. PADI4 is an epigenetic modifier, which deiminates arginine, producing the non-natural amino acid citrulline. PADI4 enhances p53 activation of a subset of target genes, which are likely to function in immune response. Y107H mice developed spontaneous tumors and metastases. The increased expression of PADI4 reduced, while knockout accelerated tumor cell growth, which is correlated with immune responses, suggesting that PADI4 contributes to tumor suppression by enhancing immune function
[145][284] (
Figure 19).
P53 activates the
Mdm2 gene and suppresses the
Arf gene to form a negative feedback loop. In addition, a new feedback loop has been identified, involving p53 inhibition of the expression of long noncoding RNA E2F1 mRNA stabilizing factor (EMS), which suppresses p53 expression
[146][285]. EMS binds to cytoplasmic polyadenylation element-binding protein 2 (CPEB2) and inhibits CPEB2 association with p53 mRNA. The disassociation of CPEB2 from p53 mRNA inhibits polyadenylation and the translation of p53 mRNA. These observations reveal a novel feedback loop between p53 and EMS, which finely regulates p53 activity. They also suggest that EMS promotes tumorigenesis, at least in part, through the negative regulation of p53 via CPEB2
[146][285].
Figure 19. Novel p53 target genes important for tumor suppression. Utilizing mouse models, novel target genes of p53, which are important for tumor suppression, have been identified. These include DNA repair gene Mlh1, cholesterol transport protein Abca1, RNA-binding protein Zmat3, phospholipid transfer protein Pltp, and epigenetic modifier Padi4.
6.2. P53 Directly Induces Apoptosis at the Mitochondria
P53 cannot induce cell cycle arrest, cellular senescence, or apoptosis in the cells obtained from triple knockout mice (p21−/−puma−/−noxa−/− mice). However, these mice are more resistant to tumorigenesis than p53-deficient mice
[135][274]. Similarly, the p53 acetylation site mutant p53(3KR) (K117R+K161R+K162R) cannot activate most p53 target genes and cannot induce cell cycle arrest, cellular senescence, or apoptosis. However, mice with p53(3KR) are also more resistant to tumorigenesis than p53-deficient mice
[147][286]. These results suggest that there are other functions of p53 that result in the induction of cell cycle arrest, cellular senescence, or apoptosis in vivo, independent of p53 target gene activation.
In addition to transcriptionally activating pro-apoptotic genes such as
Bax and
Bak, which destabilize the mitochondrial outer membrane and release cytochrome
c, p53 directly stimulates apoptosis at the mitochondria
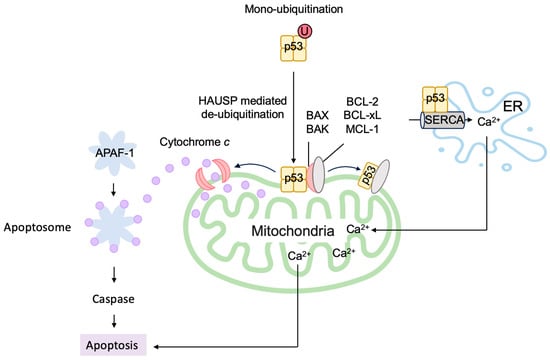
[148][287]. At the outer membrane of the mitochondria, pro-survival BCL-2 family proteins BCL-xL and BCL-2 bind and inactivate pro-apoptotic BCL-2 family proteins (multidomain and BH3-only proteins), thereby stabilizing the membrane to suppress apoptosis. A portion of p53 translocates to the mitochondria and binds to BCL-xL and BCL-2 through its DNA-binding domain, inducing conformational change and releasing pro-apoptotic BCL-2 family proteins
[149][150][151][152][153][288,289,290,291,292] (
Figure 120). Two p53 molecules form a homodimer and bind one BCL-xL molecule
[154][293]. P53 also directly activates the pro-apoptotic BCL-2 protein BAX and BAK
[151][155][290,294]. P53 also binds to pro-survival BCL-2 family protein MCL-1 and disrupts the BAK-MCL-1 complex, resulting in the release and oligomerization of BAK
[155][294] (
Figure 120). This leads to the destabilization of the mitochondrial outer membrane and release of cytochrome
c to trigger the apoptotic program.
The monoubiquitination of p53 by MDM2 facilitates translocation to the mitochondria (
Figure 120). Upon arrival at the mitochondria, p53 is deubiquitinated by mitochondrial HAUSP, leading to apoptosis
[156][295]. Hepatocyte odd protein shuttling (HOPS) binds to p53, inhibits proteasomal degradation, and facilitates the localization of p53 to the mitochondria and the induction of apoptosis
[157][158][296,297]. HOPS also interferes with the nuclear transport factor importin α to increase cytoplasmic p53 levels
[157][296].
For efficient translocation to the mitochondria and the activation of BAX, a conformational change of p53 by the prolyl isomerase Pin1 is required
[159][160][161][298,299,300], while efficient displacement of the inhibitory MCL-1 protein from BAK requires the acetylation of p53 at Lys120
[162][301]. The cytoplasmic accumulation of p53 is facilitated by FOXO3a through increasing the association of p53 with the nuclear export receptor chromosomal region maintenance 1 (CRM1)
[163][302]. Transcriptionally induced PUMA disrupts the association between p53 and BCL-xL, allowing p53 to interact with and activate BAX and BAK to promote apoptosis
[164][303].
P53 localizes to the ER and modulates Ca
2+ homeostasis. Upon genotoxic and oxidative stresses, p53 binds directly to the sarco/ER Ca
2+-ATPase (SERCA) pump in the ER, reducing the oxidation of SERCA and increasing Ca
2+ uptake into the ER. This leads to an enhanced transfer of Ca
2+ from the ER to the mitochondria, resulting in mitochondrial Ca
2+ overload and the induction of apoptosis
[165][304] (
Figure 120). Mitochondrial Lon is a multi-functional matrix protease, which associates with the Hsp60–mtHsp70 complex and shows chaperone activity. Upon oxidative stress, the level of Lon is increased and associates with p53 in the mitochondrial matrix, thereby inhibiting the apoptotic function of p53
[166][305].
Taken together, p53 directly stimulates apoptosis at the mitochondria through a variety of pleiotropic mechanisms controlled and mediated by multiple interacting factors.
Figure 120. P53 directly induces apoptosis at the mitochondria. Monoubiquitinated p53 is translocated to the mitochondria, where it is deubiquitinated by HAUSP and directly induces apoptosis. P53 binds to pro-survival BCL-2 family proteins BCL-xL and BCL-2 and releases pro-apoptotic BCL-2 family proteins. Similarly, p53 binds to pro-survival BCL-2 family protein MCL-1 and disrupts the BAK-MCL-1 complex, resulting in the release and oligomerization of BAK. P53 also directly activates the pro-apoptotic BCL-2 protein BAX and BAK. P53 binds to the SERCA pump in the ER, leading to an enhanced Ca2+ uptake and transfer to the mitochondria, which causes apoptosis.
6.3. P53 Induces Autophagy to Suppress Tumorigenesis
P53 can also induce autophagy by increasing the transcription of autophagy-related genes (
Atgs)
[161][300] (Figure 21). P53 induces the expression of damage-regulated autophagy modulator (DRAM), a lysosomal protein that facilitates autophagy. The knockdown of DRAM compromised p53-induced autophagy, indicating that DRAM is essential for p53-mediated apoptosis
[167][306]. The
Dram gene encodes a series of splice variants (SVs), of which SV4 and SV5 localize to peroxisomes and autophagosomes, respectively, and modulate autophagy
[168][307]. DRAM3 encoded by
transmembrane protein 150B (
Tmem150b) also regulates both autophagy and cell death
[169][308]. Global genomic profiling of p53-regulated genes identified a variety of autophagy-related genes, including
Atg 2,
4,
7, and
10 [170][309]. P53 induces the expression of tumor protein 53-induced nuclear protein 1 (TP53INP1), which interacts with LC3 and ATG8-family proteins and promotes autophagy-dependent cell death
[171][310]. P53 also induces the expression of cathepsin D, a major lysosomal aspartyl protease
[172][173][311,312], which contributes to autophagy
[174][313]. The inhibition of cathepsin D suppresses p53-dependent apoptosis and chemosensitivity
[172][311], suggesting a role for cathepsin D in p53-dependent cell death. Upon the depletion of growth supplements, p53 transcriptionally induces the expression of transglutaminase 2, which contributes to autophagy by facilitating autophagic protein degradation and lysosomal clearance
[175][314]. P53 causes mitochondrial defects by mitophagy and autophagic cell death by inducing the expression of DRAM and BCL-2/adenovirus E1b 19-kD protein-interacting protein 3 (BNIP-3)
[176][177][315,316]. BNIP-3-induced mitophagy may also limit the metabolic shift from mitochondrial oxidative metabolism to glycolysis by maintaining mitochondrial integrity, depending on cellular circumstances
[178][317]. Beclin-1 (ATG6) is sequestered by binding to BCL-2 and BCL-xL through its BH3 domain
[179][180][181][318,319,320]. Beclin-1 facilitates autophagy by interacting with the autophagy-related gene products. Thus, it is expected that the induction of BH3-only proteins by p53 releases Beclin-1 from BCL-2 and BCL-xL by competing with their binding to Beclin’s BH3 domain, thereby facilitating autophagy.