Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Michael John Tolentino.
Age-related macular degeneration (AMD) is an age-related condition that progressively impairs central vision with increasing age. AMD affects the central portion of the retina called the macula, which is required for central vision and visually demanding tasks like recognizing faces, reading, and driving.
- microglia
- macrophages
- macular degeneration
- sialic acid
- Siglecs
- nanoparticles
1. Age-Related Macular Degeneration (AMD)
1.1. Background
AMD is an age-related condition that progressively impairs central vision with increasing age. AMD affects the central portion of the retina called the macula, which is required for central vision and visually demanding tasks like recognizing faces, reading, and driving. Because central vision is important for these higher tasks, the impairment brought on by AMD results in decreased independence, mobility, and quality of life [1]. The potential for this form of vision loss is rated by surveyed individuals as one of the worst health outcomes possible [2].
AMD can be classified into early, intermediate, and late stages [3]. It is a disease that affects persons over the age of 60, and its incidence is increasing due to the rapid growth in the elderly population worldwide [4]. The prevalence of early- to intermediate-stage AMD in the US in 2019 reached approximately 18.34 million people [5]. This prevalence represents three times the number of patients with Alzheimer’s disease and is equal to all patients with a cancer diagnosis excluding melanoma [6,7][6][7]. By 2040, this disease that progressively causes central visual loss is estimated to affect 288 million people worldwide [8]. Currently, macular degeneration represents the third cause of vision loss secondary to ocular pathology worldwide [9]. Furthermore, the disease burden disproportionately affects less developed and low-income countries in Africa and Asia [10,11][10][11].
1.2. Clinical Presentation
The hallmark of early-stage AMD is the development of yellow subretinal deposits on the macula called drusen and/or abnormal pigmentary change called pigmentary mottling, clumping, or retinal pigment epithelial (RPE) change. When these changes worsen and reach a certain density, the stage is classified as intermediate. The late stage is reached when geographic atrophy (GA), neovascularization, or both develop [3].
From a visual function perspective, AMD can be divided into vision-threatening and non-vision-threatening stages. While drusen and RPE changes constitute the initiation of clinically detectable disease, these findings are usually not accompanied by any symptoms or noticeable vision loss [12]. Most patients with drusen and RPE changes alone do not develop significant vision loss. On the other hand, most patients progressing to the late-stage complications of geographic atrophy, exudative macular degeneration, or both will eventually develop moderate to severe central vision loss [13]. The prevalence of late-stage AMD in the US in 2019 was calculated to be 1.49 million people, which represents 7.5% of all the patients with AMD [5]. This difference between the prevalence of the non-vision-threatening early stage and vision-threatening late stage indicates that most patients will not develop moderate to severe central visual loss.
Macular degeneration is analogous to cardiovascular disease. Drusen and RPE changes are synonymous with plaque buildup on the coronary artery walls. As the drusen and RPE changes progress to intermediate and advanced stages, the risk of developing wet AMD or geographic atrophy increase [14]. In heart disease, advancing coronary artery plaque buildup results in coronary artery stenosis, which could lead to myocardial infarction, synonymous with wet macular degeneration, or ischemic cardiac heart failure, synonymous with geographic atrophy. Like cardiovascular disease, high cholesterol and coronary artery disease do not invariably lead to heart attack or cardiac heart failure.
Development of drusen and RPE changes, like plaque buildup and high cholesterol, are insidious and asymptomatic. These signs are predominantly detected by physician examination or sophisticated imaging tools such as optical coherence tomography (OCT) [15]. The initial development of exudative macular degeneration or geographic atrophy are often symptomatically undetectable, like ischemic heart disease or silent myocardial infarction, and require medical examination or sophisticated imaging technology to detect, such as wide-field ophthalmoscopy [16]. Exudative AMD often presents with mild visual symptoms such as metamorphopsia, distorted vision, color change, contrast abnormality, or mild visual loss [17]. In GA, the location of atrophy determines the severity of vision loss, where central-involving GA will result in the recognition of a blind spot while non-central-involving GA will be asymptomatic [18,19][18][19].
1.3. Risk Factors
Risk factors that have been consistently associated with AMD are age, ethnicity, smoking, and genetic polymorphisms. Age is the most important risk factor for the development and progression of both early- and late-stage AMD. This importance is demonstrated by the low 3.5% prevalence of early and 0.1% for late AMD in those younger than 59 and the high 17.5% prevalence of early and 9.8% for late AMD in those older than 85. This difference is a 5 (early) and 98 (late) times increase in prevalence between these two age groups [20]. In regard to ethnicity, white Europeans have the highest annual incidence of both early and late AMD [21]. Smoking is the strongest associated modifiable risk factor in both early- and late-stage AMD patients [22].
In total, 103 AMD genes have been associated with AMD, but the most significantly associated are the complement factor H (CFH), age-related maculopathy susceptibility 2/high-temperature requirement A serine peptidase 1 (ARMS2/HTRA1), and apolipoprotein E (APOE) polymorphisms [23]. The most associated polymorphism to AMD is found in the CFH loci: the substitution of the histidine for tyrosine at the 42 codon of chromosome 1-region 31 (rs1061170), which results in the alteration of the sialic acid/heparin binding site in the short consensus repeat region 7 of the CFH protein [24,25,26][24][25][26]. Effectively, this polymorphism reduces binding with self-associated molecular patterns (SAMPs) and permits unchecked alternative complement activation and resultant chronic immune activation [27,28,29,30][27][28][29][30].
ARMS2/HTRA1 are genes that exhibit a high degree of linkage disequilibrium, so it is difficult to determine which polymorphism is responsible for its association with AMD [31]. ARMS2 is a protein that is expressed by human monocytes, binds to apoptotic cells, and recruits properdin, which facilitates C3b opsonization and phagocytosis of apoptotic and necrotic cells [32]. The polymorphism in ARMS2 reduces phagocytosis of necrotic and apoptotic debris and may result in accumulation of drusen material. The absence of ARMS2 can result in both reduced phagocytosis of apoptotic cells and increased nonspecific phagocytosis of healthy cells, leading to cellular loss like geographic atrophy [32].
HTRA1 polymorphism results in increased production of the HTRA1 protein, which is a serine peptidase known to cleave APOE [33]. Increased cleavage would inactivate APOE, mimicking the dysfunction brought about by the polymorphisms in APOE found in AMD. APOE’s best-known role is to regulate the transportation of lipids and cholesterol in the retina and brain [34]. Another major role is to protect lipids from complement attack by binding and activating CFH and protecting high-density lipoproteins (HDLs) from complement attack [35]. Polymorphisms in APOE associated with AMD also reduce APOE levels, dysregulating lipid and cholesterol clearance and inciting inflammation by not protecting the lipids from complement attack [35].
2. Central Role of Inflammation and Parainflammation in AMD
2.1. Clinical Evidence of Inflammation
In the early and intermediate stages of macular degeneration, measurable photoreceptor dysfunction is reflected in abnormalities dark adaptation, visual field, photo stress, and electro-retinographic changes [36,37,38,39][36][37][38][39]. These disease-correlated changes in visual function demonstrate inflammation as the underlying pathology behind all stages of macular degeneration. Prolonged dark adaptation, which progressively worsens in step with stage of AMD, is caused by inflammatory visual cycle impairment and indicates a worsening of inflammation as AMD progresses [40,41][40][41]. In one study, patients with early AMD had qualitative visual changes and symptoms of distortion that could be detected and quantified by visual field analysis [37]. These deficits of form recognition and sensitivity were found in areas of RPE atrophy not defined as geographic atrophy within this patient population, indicating asymptomatic vision loss in areas of overactive inflammation [37]. Photo stress recovery time, a measure of visual pigment recycling time, was inversely correlated with visual acuity, and inflammation-induced prolongation of recovery time was directly correlated with the presence or absence of geographic atrophy and advancing age [39]. In late AMD, inflammatory slowing of implicit time and amplitude reduction on electroretinograms (ERGs) of patients with geographic atrophy were seen in areas bordering fundus autofluorescence-defined geographic atrophy [42]. Foveal ERG performed in fellow eyes of patients with wet AMD who did not have severe visual acuity changes demonstrated implicit time prolongation, indicating that fellow eyes were also undergoing inflammation [38]. Visual cycle alteration represented the first indication of the central role of inflammation in AMD.2.2. Anti-Oxidant Therapy for Early-Stage AMD
While clinical findings in AMD pointed to inflammation as the central driver of AMD progression, initial therapeutics for AMD were focused on anti-oxidation. To this day, the only consensus treatment for early/intermediate-stage AMD is the use of anti-oxidant therapies studied in a large National Eye Institute study called the Age-Related Eye Disease Study 1 and 2 (AREDS 1 and 2) [43]. This study has produced over 28 reports and countless publications on the benefit of anti-oxidant therapy for the prevention of late AMD progression [44]. While the conclusions from these studies demonstrated a reduction in the rate of development of large drusen, geographic atrophy, and exudative AMD, the modest reduction in rate of progression implicates oxidation as only a stimulator of inflammation rather than the main driver for AMD progression [45,46][45][46].2.3. Oxidation-Induced Dysfunctional Parainflammation in Early AMD
Demographic, environmental, and genomic risk factors combined with electrophysiological and psychophysical studies definitively implicate retinal inflammation as the major underlying factor in the development of all stages of AMD [47,48,49,50][47][48][49][50]. The degree of dysfunctional inflammation determines the stage and clinical presentation [51]. Multiple papers have described macular degeneration as a disease that is initiated by dysregulated parainflammation leading to the drusen stage of AMD then progressing to overt inflammation that triggers the late stage of AMD [52,53,54,55][52][53][54][55]. In 2008, Medzhitov postulated a parainflammatory state that lay between basal homeostatic conditions and true inflammation [56]. This parainflammatory state is considered an adaptive immune response to low level tissue stress such as the age-related accumulation of oxidative byproducts [57]. This acquired dysfunctional parainflammation that occurs with aging has been termed inflamaging and likely explains the early stages of AMD with drusen development and RPE changes [58]. The development of late exudative and geographic atrophy stages, on the other hand, is an overt innate immune activation with end-stage pathology determined by macrophage polarization [59]. Oxidative damage is the main tissue stress that stimulates parainflammation in the retina [60] (Figure 1(1)). The macula, which is exposed to photo, metabolic, phagocytic, and mitochondrial reactive oxygen species (ROS) production, is a site of tremendous oxidative stress. An association between blue light exposure (photo-oxidative light) and low anti-oxidant levels demonstrated an association with early and neovascular forms of AMD, implicating photo-oxidative light as a contributor to oxidative stress in AMD [61].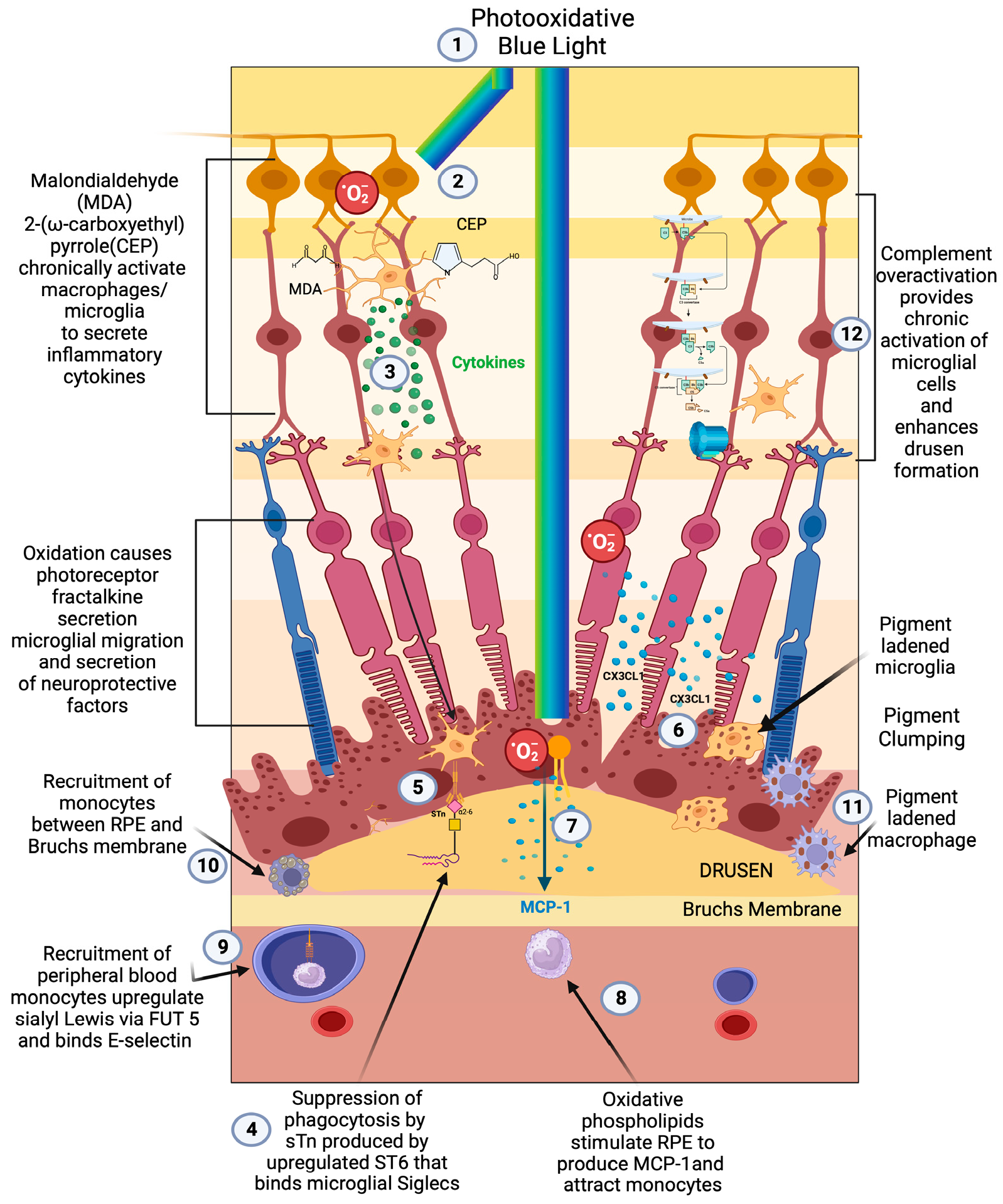
Figure 1. Early/intermediate-stage AMD. (1) Photo-oxidative blue light (2) oxidizes retinal lipids to produces oxidative byproducts (MDA, CEP) and reactive oxygen species, (3) which activate microglial cells to secrete cytokines. These activated macrophages are hindered from becoming phagocytic due to the (4) upregulation of ST6 that produces (5) sTN on the surface of retinal cells and drusen that agonizes Siglecs to prevent phagocytosis of drusen. (6) Reactive oxygen species also cause photoreceptors to secrete CX3CL1, which promotes migration of microglia and macrophages to the retina. (7) Oxidized phospholipids stimulate RPE cells to produce MCP—1 (8), which recruits peripheral blood monocytes (PBMCs) to areas of phospholipid oxidation. (9) Upregulation of FUT5 on PBMCs localizes monocytes to areas that are secreting chemokines such as CX3CL1 that upregulate e-selectin on vascular endothelium. (10) Monocytes are found between RPE cells and Bruch’s membrane. (11) Microglial cells and macrophages are also found to be pigment-ladened in this sub-RPE space and at the RPE cell layer, which appears as RPE pigment clumping. (12) The overactivation of complement caused by the polymorphism-induced impaired function of CFH will produce complement pathway metabolites that accumulate in drusen and can activate microglial cells.
2.4. Complement Pathway-Induced Parainflammation
Despite the strong association of complement factor polymorphisms with the development of AMD and the development of two FDA-approved treatments for geographic atrophy (Pegcetacoplan and Avacincaptad pegol), there is a consensus that complement does not fully account for disease development [82,83,84,85][82][83][84][85]. Its direct role in disease may be in the development of drusen, and it may play only an indirect role in the development of late-stage disease [86] (Figure 1(12)). Evidence to support this role in producing drusen is found in the proteomic analysis of drusen from retinas of patients with AMD, where a large proportion of patients had complement proteins 9 (C9) and 3(C3) in their drusen [87]. C9 is a protein found in conjunction with the c5-9 complex or membrane attack complex. This finding agreed with earlier histopathologic analysis that corroborated the presence of multiple complement factors in drusen [88]. A meta-analysis found that systemic complement overactivation was a feature associated with early/intermediate AMD rather than late-stage geographic atrophy [89]. While no trials have been performed to determine the effect of complement factor depletion on the development of drusen, several trials have looked at the effect of complement C3(Pegcetacoplan) and C5 (Avacincaptad pegol) depletion and demonstrated a modest reduction in the growth rate of GA [90,91][90][91]. According to both pre-clinical and clinical studies, complement overactivation is important in initiating AMD by exacerbating parainflammatory overactivation and dysfunction, which enhances drusen formation. The inability of profound complement depletion to halt the progression of late-stage GA relegates complement as a minor player in the pathogenesis of late-stage AMD [90,91][90][91].2.5. Early-Stage AMD Dysfunctional Parainflammation
At the cellular level, the early/intermediate stage is the accumulation of oxidized, metabolic, inflammatory debris that appears as yellow sub retinal deposits called drusen [87]. The pigmentary clumping seen in AMD represents pigment-ladened microglia or macrophage migration towards the retina, Bruch’s membrane, and under surface of the RPE [92] (Figure 1(9–11)). The accumulation of proinflammatory oxidative byproducts CEP and MDA leads to phagocytic/cytokine secreting microglial polarization, activation of parainflammatory mechanisms, and, potentially, recruitment of peripheral blood macrophages to the subretinal space to compensate for this accumulation of subretinal toxic substances [55] (Figure 1(2,3,10)). With age-dependent RPE cell senescence and dysfunction of autophagy, parainflammatory activation of microglial cells accelerates, resulting in the recruitment of peripheral blood-derived macrophages into the subretinal space [93].2.6. Microglia’s Central Role in Dysfunctional Parainflammation
Microglia play a central role in modulating parainflammation [94] (Figure 2(1)). Microglial overactivation represents a common pathomechanism in a variety of retinal degenerative diseases and is often overactivated prior to the onset of overt retinal cell death [95]. In the retina, microglia’s dynamic motility allows comprehensive surveillance coverage of the entire retina in a short time period [96] (Figure 2(5)). This motility allows microglia to interact with retinal neurons and macroglia and play a central role in retinal homeostatic maintenance and clearance of cellular and metabolic debris [97].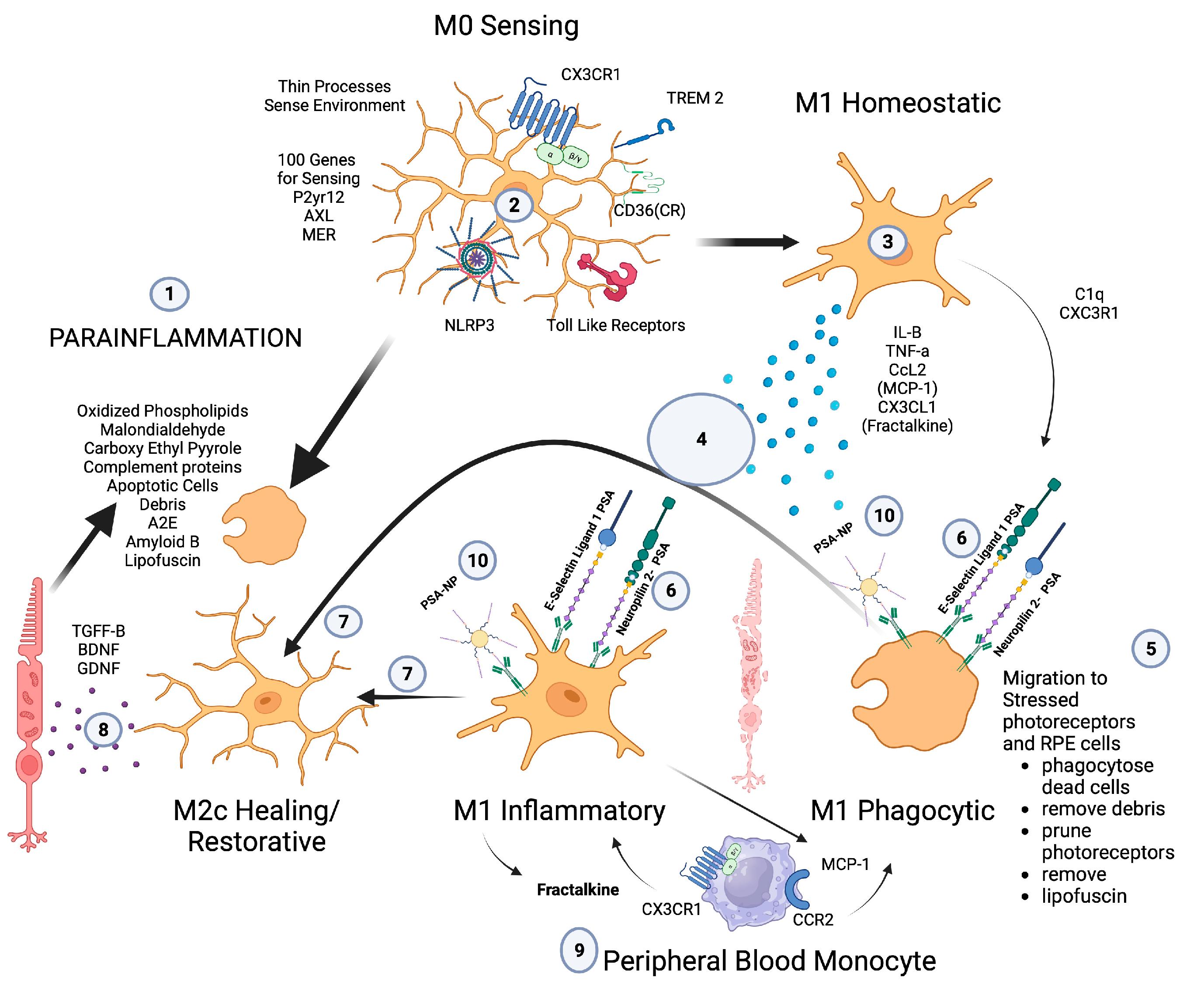
Figure 2. Microglial parainflammatory regulation: Microglia remove toxic metabolites, apoptotic cells, and oxidative debris while maintaining the health of photoreceptors and retinal cells in a process called parainflammation. (1) Parainflammation is initiated by pattern recognition receptors found on (2) M0 microglia such as TLR, TREM2, CX3CR1, NLRP3, and CD36, which use these receptors to bind and sense toxins, apoptotic cells, damage, and pathogen-associated molecular patterns. Once microglia are activated, they polarize to the (3) M1 like state, (4) secrete proinflammatory cytokines, and (5) migrate to the areas of stressed RPE cells and photoreceptors. (6) Upon activation, neuropilin-2-PSA and E-selectin ligand 1-PSA are secreted by these activated microglia. (6) The PSA on these glycoproteins binds Siglecs on activated microglial cells (7) to polarize them into the M2c healing state (8), which releases growth factors to protect and regenerate the stressed photoreceptors and RPE cells. (9) This homeostatic maintenance function of microglial parainflammation, if not adequately modulated with sialic acid checkpoint regulation, will result in recruitment of peripheral blood monocytes and AMD disease progression. (10) A PSA-nanoparticle (PSA-NP) mimics E-selectin ligand 1 PSA and Neuropilin 2-PSA to (7) polarize M1 activated microglia into the M2c healing state.
2.7. Peripheral Blood-Derived Macrophages’ Role in Transition to Late AMD
The major role of peripheral blood monocyte-derived macrophages in the progression of the early drusen stage to late-stage geographic atrophy and exudative AMD is evidenced by the increase in number of activated macrophages in the choroid and Bruch’s membrane as AMD progresses. The observation that the highest number of activated macrophages are found in eyes with choroidal neovascularization further supports this central role [102]. The recruitment of peripheral blood-derived macrophages is a major function of retinal microglial cells when injury or toxic material overwhelm the microglia’s ability to phagocytose toxic oxidative byproducts, injured cells, or apoptotic cells [103]. To maintain macular health, the microglia must phagocytose oxidative byproducts (oxidized phospholipids, malondialdehyde, carboxyethyl pyrrole), apoptotic cells, complement proteins, abnormal proteins (Amyloid B), and toxic metabolites (A2E). If microglia do not encounter appropriate checkpoint ligands in the form of sialic acid, then chemokine signaling will recruit peripheral blood macrophages [104] (Figure 2(9)). Patho-mechanistically, the recruitment of peripheral blood monocytes in clinically evident macular degeneration is mediated by microglial chemokine signaling, which is responsible for the recruitment of monocytes to the choroid and the retina. The chemokine receptors CCR2 and CX3CR1 and their respective ligands CCL2 (monocyte chemotactic protein-1, MCP-1) and CX3CL1 (fractalkine) mediate this recruitment [68,105,106][68][105][106]. While fractalkine-CX3CR1 signaling is proinflammatory, it is also critical for progesterone-mediated neuroprotection of the retina [107]. This dual role of Fractalkine-CX3CR1 interaction in microglial proinflammatory activation and neuroprotective properties demonstrates the necessity of tight regulation of microglial cells. If the proinflammatory properties of fractalkine-CX3CR1-activated microglia could be checked, but they could maintain their neuroprotective properties, then polarizing macrophages to the resolution state would eliminate inflammation and attenuate phagocytosis while providing neuroprotection. Without appropriate checkpoint regulation of microglial cells, the inflammatory activation state of microglia will promote more photoreceptor degeneration rather than rescue of photoreceptors.2.8. Macrophage Recruitment Indicator of Late-Stage AMD
The central role of macrophages in the pathogenesis of neovascular wet AMD is widely accepted [108[108][109][110][111],109,110,111], but the critical role of activated macrophages in the pathogenesis of geographic atrophy has been widely overlooked due to the focus on the complement pathway [112]. Prior to the genomic association between complement factors and AMD, histopathologic evidence pointed to macrophage/mononuclear phagocyte/multinucleated giant cells as the central causative factor in the pathogenesis of GA and the main phagocytic cause of retinal cellular clearance that manifests as RPE and photoreceptor loss [101,113][101][113] (Figure 3(5,6,9)).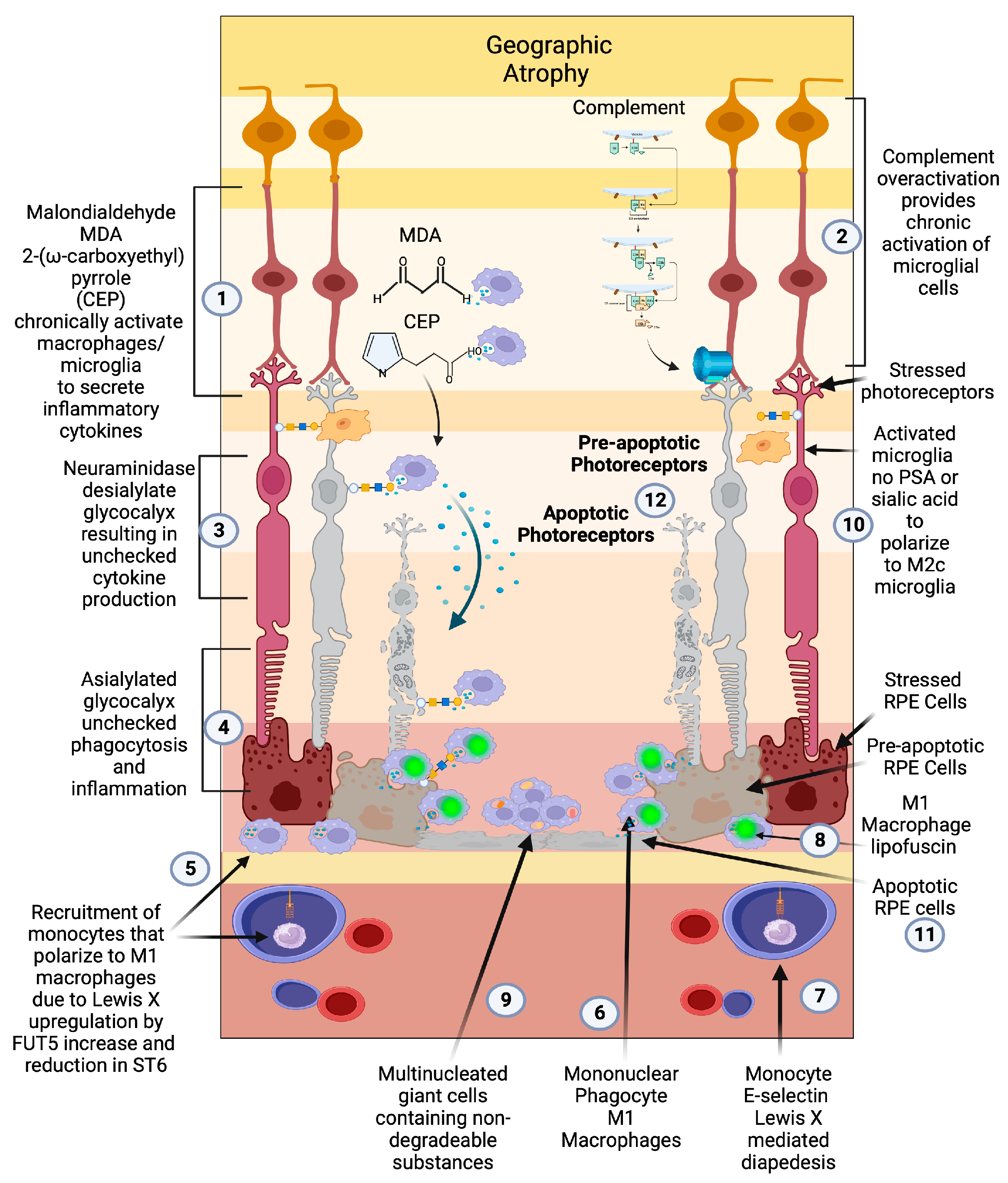
Figure 3. Geographic atrophy: (1) Progressive accumulation of oxidative byproducts (CEP, MDA) and (2) chronic overactivated complement pathway chronically activate microglia (3), which secrete neuraminidase and desialylate photoreceptors and RPE cells. (4) This loss of sialylation prevents restoration of homeostasis. (5) Chronically overactivated phagocytosis and inflammation recruits peripheral blood macrophages by upregulation of the fucosyltransferase FUT5 that produces Lewis X glycosylation on monocytes to bind E-selectin and (6) localize monocytes to sites of inflammation (7), allowing for diapedesis of the monocyte across the blood–retinal barrier. (8) These monocytes polarize to M1 macrophages when they enter the retina and are not able to clear substances like lipofuscin and other undegradable substances. (9) The macrophages form multinucleated giant cells because they phagocytose structures that are undegradable. (10) Since there is no sialic acid or polysialic acid to polarize to the healing M2c state, the macrophages are unchecked and result in elimination (11) first of the RPE cells then the (12) photoreceptors. The unchecked macrophages are the main determinant of growth of geographic atrophy.
2.9. Macrophage Polarization Determines Late-Stage AMD
The classically activated (M1) and alternatively activated (M2) binary description of macrophage polarization, based on biomarker expression and cytokine production, does not reflect the true character of these subtypes. A function-based description of macrophage polarization better characterizes their role in pathology [59,117,118][59][117][118]. The M1 polarization state is characterized as the proinflammatory phagocytic state. The M2 state can be subdivided into four M2 subtypes. The M2 a, b subtype are the anti-inflammatory pro-fibrotic type, the M2d is the pro angiogenic phenotype, and the M2c is the anti-inflammatory and neuroprotective type [117,118,119][117][118][119]. The M2c can also dedifferentiate myofibroblasts, so is considered anti-fibrotic [120]. In vitro, M1 macrophages are predominantly neurotoxic with modest axon growth-promoting effect, in contrast to the M2 macrophages, which promote long-distance axon growth without neurotoxicity [121]. In vivo studies in traumatic brain or spinal cord injury have characterized the time course and characteristics of M1/M2 polarization after injury [121,122][121][122]. M1-like macrophages release oxidative metabolites and proteases that kill neurons and glial cells [121]. In contrast, M2-like cells facilitate tissue repair [123]. In spinal cord injury models, increased M2c microglia expressed in the first week after injury correlated with better neurological outcome, indicating a healing neuroprotective function of M2c microglia/macrophages [122]. A time course study comparing M1 versus M2 levels in this model show that M1 expression is upregulated for at least a month, while M2 levels diminish drastically 1 week post injury. The level of M2a and M2c upregulation within the first week correlated well with neurological recovery. The neurological recovery resulted from the neuroprotective properties of microglia polarized to the M2c or M2a state. These properties were the secretion of trophic factors such as brain-derived neurotrophic factor (BDNF) and glial-derived neurotrophic factor (GDNF) and the ability to perform controlled phagocytosis, which prevents necrosis of surrounding tissue [124] (Figure 2(8)). While at first glance, macular degeneration may not appear to be a disease of acute CNS injury, the macrophage microglial behavior in the retina is consistent with what is seen in injury models of the CNS [55,125][55][125]. Eyes with advanced AMD had a higher ratio of M1 to M2 macrophages than age-matched normal autopsied eyes [126]. This enhanced chronic expression of neurotoxic phagocytic M1 macrophage with the reduction in M2c neuroprotective macrophages results in unchecked photoreceptor, RPE cell degeneration, and phagocytosis (geographic atrophy) [126]. The abundance of M1 polarized macrophages and adenosine polarize M1 to M2D VEGF-producing macrophages, resulting in the development of subretinal neovascularization [127]. If the M2 a, b polarization predominates, then retinal fibrosis will develop (disciform scar) [59]. Like CNS injury, the failure to polarize M1 macrophages towards the M2 c state will result in failure of functional recovery [122] (Figure 4).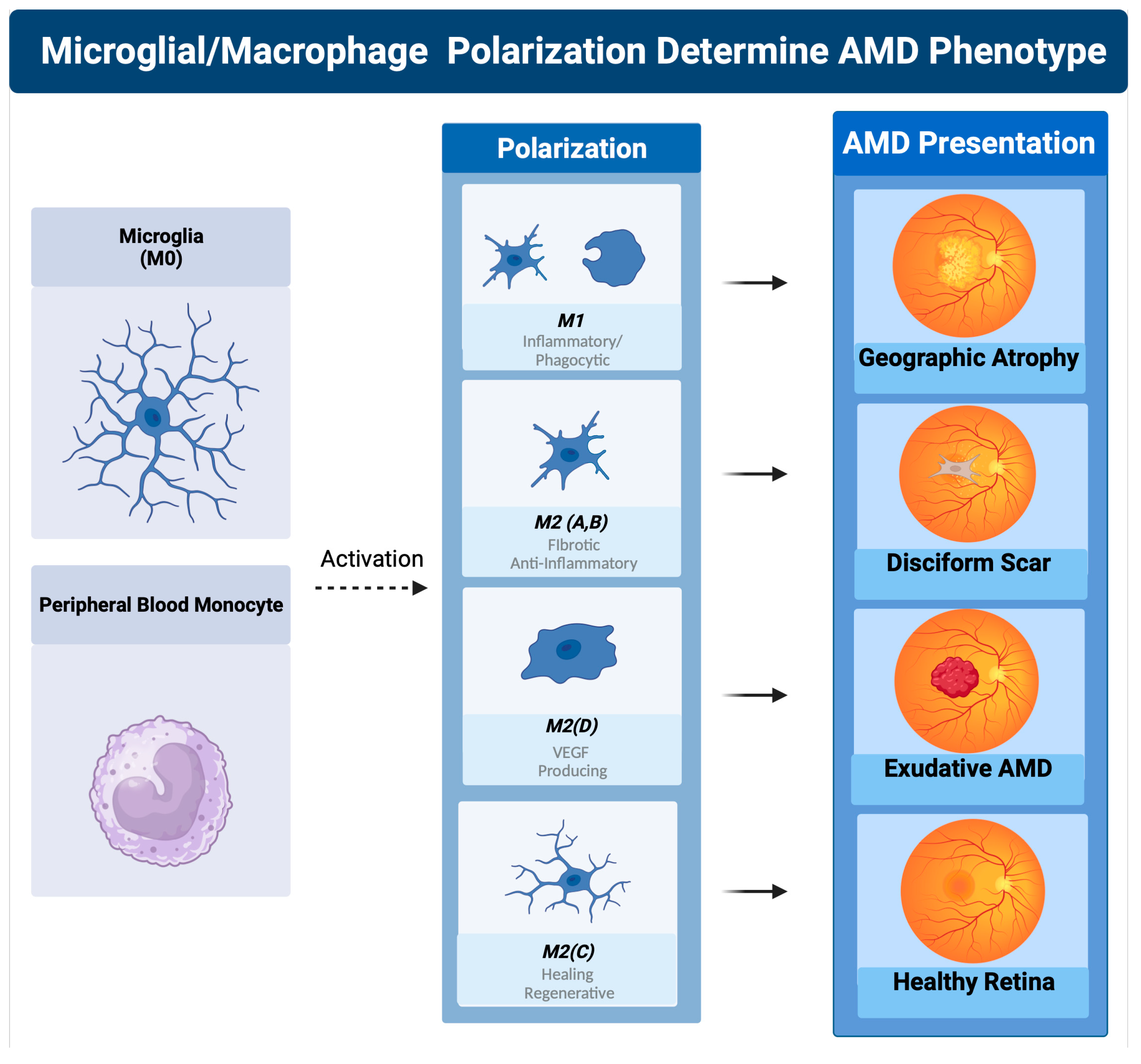
Figure 4. Macrophage polarization determines AMD phenotype. The plasticity and different polarization states correlate with the clinical picture seen in late-stage macular degeneration. In geographic atrophy, RPE cells and photoreceptors are phagocytosed as a function of the M1 polarized phagocytic macrophages. Exudative AMD is neovascularization produced by overexpression of VEGF, the main cytokine secreted by the M2d polarization state. The sequelae of exudative AMD untreated with anti-VEGF therapies is a disciform fibrotic scar in the control of the pro-fibrotic M2 A, B state. With appropriate sialic acid signaling such as PSA, all polarization states can transform into the healing M2c state.
2.10. Sialylation Controls Microglial Activation and Macrophage Polarization in AMD
Non-genomic factors determine microglial activation and macrophage polarization in AMD. To determine which of these factors were important in AMD, Emilsson et al. performed a large proteo/genomic analysis of serum proteins in patients with macular degeneration [128]. This group identified several serum proteins that were elevated differentially in early and late AMD. As expected, serum protein complement factor H related 1 (CFHR1) correlated with early and late AMD. Complement factor H related 5 (CFHR5) correlated only with late-stage AMD. A novel but important finding from this study was the differential correlation of a class of proteins called sialyltransferase with different stages of AMD [128]. In this analysis, elevated serum protein levels of Alpha-N-acetylgalactosaminide alpha-2,6-sialyltransferase 1 (ST6GALNAC1/ST6) and Alpha-(1,3)-fucosyltransferase (FUT5) were highly correlated with patients with early-stage AMD [128]. In late-stage AMD patients, only FUT5 was found to be highly correlated. The expression of FUT5 in both late and early AMD and the expression of ST6 in only early AMD point to alterations of sialic acid glycan expression as a major determinant in the progression to late-stage AMD. It also implicates the absence of sialic acid self-associated molecular patterns (sSAMPs) in late AMD, which bind sialic acid-binding immunoglobulin-like lectins (Siglecs) to resolve activated macrophages to the resolution M2c state [129]. The loss of ST6 expression and the upregulation of FUT 5 in late AMD explain why microglia and macrophage activation is unchecked. When ST6 is expressed, the sialylated Tn antigen (sTn) rapidly sialylates the inflamed or oxidatively damaged glycocalyx of cells as well as the debris of these damaged cells [130]. This sialylation of the debris found in drusen [131] by sTN resolves activation of microglial cells, resulting in reduced phagocytosis, no recruitment of peripheral blood macrophages, no overt inflammation, and eventual accumulation of metabolic and inflammatory debris (Figure 1(9)). This debris, combined with complement pathway-created proteins and lipid byproducts, will accumulate in the subretinal space and worsen drusen and pigmentary clumping [88]. During the lates stage of AMD, ST6 is no longer upregulated; instead, FUT5 becomes the predominant sialyltransferase. FUT5 is a critical glycotransferase that produces Lewis x (3Gal𝛽�1,4[Fuc𝛼�1,3] GlcNAc-), sialyl Lewis x (sLex,1 NeuAc𝛼�2, 3Gal𝛽�1,4[Fuc𝛼�1,3]GlcNAc-), Lewis a (Lea, 3Gal𝛽�1,3[Fuc𝛼�1,4]GlcNAc), sialyl Lewis a (sLea, NeuAc𝛼�2, 3Gal𝛽�1,3[Fuc𝛼�1,4]GlcNAc-), and Lewis b (Leb, Fuc 𝛼�1,2Gal𝛽�1,3[Fuc𝛼�1,4]GlcNAc-). These glycans are the main binding determinants for selectins, in particular, e-selectin, which is responsible for localizing monocytes to areas of active inflammation [132]. Without sialic acid SAMP checkpoint restraint on activated microglia and recruited macrophages, immune cell activation will overproduce cytokines such as VEGF and phagocytose both drusen and underlying RPE cells and overlying photoreceptors, resulting in geographic atrophy (Figure 3(4,5)). Clinical and proteomic findings in all stages of macular degeneration point to loss of microglia and macrophage sialic acid/Siglec immune checkpoint control as a major determinant of disease progression to late-stage geographic atrophy or CNV [133,134][133][134]. If appropriate sialic acid-mediated checkpoint control could be regained and homeostatic neuroprotective microglial function restored, late-stage disease could be halted and inflammation-impaired visual function could potentially be improved [135]. The alteration of the glycocalyx, “the sugar coat” of retinal cells, which in healthy young retinas are decorated with sialic acid caps (SAMPs), which agonize Siglec checkpoint receptors, determines the activation and polarization state of microglia and macrophages. By either binding to complement factors such as CFH and properdin or to Siglec receptors on immune cells, these sialic acid caps serve as checkpoint ligands to resolve innate immune activation [104]. This overlooked sialic acid sugar coating that determines the immune self is the master checkpoint regulator of immune cells and immune function [136]. Even in a non-inflammatory environment, the sugar coating on immune and host cells determines the polarization state of microglia and macrophages [137]. If there are enough sialic SAMP patterns, microglia are placed into the resolved state, which allows them to take on a neuroprotective and regenerative phenotype by producing cytokines such as BDNF or CTGF and selectively pruning neuronal dendrites and apoptotic cells [118,138][118][138]. In this sialic SAMP-checked state, microglia also can sense stressed, damaged, and sick cells or those undergoing apoptosis, which activate the microglia to the transient M1-like state to eliminate the damaged cell-released toxins and apoptotic cells [139]. If there are adequate sialic SAMPs available to bind the microglial Siglecs, the activation is short-lived and will result in the repolarizing of these M1 microglia to the M2c-like state [118]. Macrophage polarization determines how macular degeneration will clinically manifest, but it is the loss of cell surface sialic acid SAMP, a glycoimmune checkpoint ligand, that permits the disease to transition between the different polarization states [59,140][59][140]. Over time, overactivated complement and cumulative oxidative damage erodes the complex sialylated glycan structures of the cellular glycome [141]. Loss of sialic acid glyco-immune checkpoint restraint on microglial cells permits M1 activation of the microglia and active recruitment of peripheral blood macrophages. FUT5-dependent production of Lewis antigens drives diapedesis of peripheral blood monocytes by binding e-selectin on the inflamed vascular endothelium [132] (Figure 3(5,7)). The recruitment of predominantly M1 polarized macrophages to the retina in the absence of appropriate sialic acid SAMPs results in unchecked inflammation and clinical progression of macular degeneration. A potential therapy would be to agonize the Siglec and CFH checkpoint receptors with a sialic acid SAMP mimetic to attenuate complement amplification and repolarize activated microglia/macrophages to their non inflammatory, healing, and homeostatic form. This therapeutic strategy would normalize checkpoint control of the immune cell, in contrast to other anti-inflammatory strategies that deplete cytokines to inhibit inflammatory pathways [59,140][59][140].References
- Taylor, D.J.; Hobby, A.E.; Binns, A.M.; Crabb, D.P. How does age-related macular degeneration affect real-world visual ability and quality of life? A systematic review. BMJ Open 2016, 6, e011504.
- Scott, A.W.; Bressler, N.M.; Ffolkes, S.; Wittenborn, J.S.; Jorkasky, J. Public Attitudes About Eye and Vision Health. JAMA Ophthalmol. 2016, 134, 1111–1118.
- Keenan, T.D.L.; Cukras, C.A.; Chew, E.Y. Age-Related Macular Degeneration: Epidemiology and Clinical Aspects. Adv. Exp. Med. Biol. 2021, 1256, 1–31.
- Izekenova, A.K.; Kumar, A.B.; Abikulova, A.K.; Izekenova, A.K. Trends in ageing of the population and the life expectancy after retirement: A comparative country-based analysis. J. Res. Med. Sci. 2015, 20, 250–252.
- Rein, D.B.; Wittenborn, J.S.; Burke-Conte, Z.; Gulia, R.; Robalik, T.; Ehrlich, J.R.; Lundeen, E.A.; Flaxman, A.D. Prevalence of Age-Related Macular Degeneration in the US in 2019. JAMA Ophthalmol. 2022, 140, 1202–1208.
- 2021 Alzheimer’s disease facts and figures. Alzheimers Dement 2021, 17, 327–406.
- Global Burden of Disease Cancer, C.; Kocarnik, J.M.; Compton, K.; Dean, F.E.; Fu, W.; Gaw, B.L.; Harvey, J.D.; Henrikson, H.J.; Lu, D.; Pennini, A.; et al. Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life Years for 29 Cancer Groups From 2010 to 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. JAMA Oncol. 2022, 8, 420–444.
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116.
- Blindness, G.B.D.; Vision Impairment, C.; Vision Loss Expert Group of the Global Burden of Disease Study. Causes of blindness and vision impairment in 2020 and trends over 30 years, and prevalence of avoidable blindness in relation to VISION 2020: The Right to Sight: An analysis for the Global Burden of Disease Study. Lancet Glob. Health 2021, 9, e144–e160.
- Zou, M.; Zhang, Y.; Chen, A.; Young, C.A.; Li, Y.; Zheng, D.; Jin, G. Variations and trends in global disease burden of age-related macular degeneration: 1990-2017. Acta Ophthalmol. 2021, 99, e330–e335.
- Xu, X.; Wu, J.; Yu, X.; Tang, Y.; Tang, X.; Shentu, X. Regional differences in the global burden of age-related macular degeneration. BMC Public Health 2020, 20, 410.
- Pinelli, R.; Bertelli, M.; Scaffidi, E.; Fulceri, F.; Busceti, C.L.; Biagioni, F.; Fornai, F. Measurement of drusen and their correlation with visual symptoms in patients affected by age-related macular degeneration. Arch. Ital. Biol. 2020, 158, 82–104.
- Schultz, N.M.; Bhardwaj, S.; Barclay, C.; Gaspar, L.; Schwartz, J. Global Burden of Dry Age-Related Macular Degeneration: A Targeted Literature Review. Clin. Ther. 2021, 43, 1792–1818.
- Amarasekera, S.; Samanta, A.; Jhingan, M.; Arora, S.; Singh, S.; Tucci, D.; Lupidi, M.; Chhablani, J.; Age Related Macular Degeneration study, g. Optical coherence tomography predictors of progression of non-exudative age-related macular degeneration to advanced atrophic and exudative disease. Graefes Arch. Clin. Exp. Ophthalmol. 2022, 260, 737–746.
- Liu, J.; Laiginhas, R.; Shen, M.; Shi, Y.; Li, J.; Trivizki, O.; Waheed, N.K.; Gregori, G.; Rosenfeld, P.J. Multimodal Imaging and En Face OCT Detection of Calcified Drusen in Eyes with Age-Related Macular Degeneration. Ophthalmol. Sci. 2022, 2, 100162.
- Matsuba, S.; Tabuchi, H.; Ohsugi, H.; Enno, H.; Ishitobi, N.; Masumoto, H.; Kiuchi, Y. Accuracy of ultra-wide-field fundus ophthalmoscopy-assisted deep learning, a machine-learning technology, for detecting age-related macular degeneration. Int. Ophthalmol. 2019, 39, 1269–1275.
- Papadopoulos, Z. Recent Developments in the Treatment of Wet Age-related Macular Degeneration. Curr. Med. Sci. 2020, 40, 851–857.
- Sunness, J.S. What you see is not always what you get in atrophic macular disease. Retin. Cases Brief. Rep. 2008, 2, 205–208.
- Sunness, J.S.; Rubin, G.S.; Zuckerbrod, A.; Applegate, C.A. Foveal-Sparing Scotomas in Advanced Dry Age-Related Macular Degeneration. J. Vis. Impair. Blind. 2008, 102, 600–610.
- Colijn, J.M.; Buitendijk, G.H.S.; Prokofyeva, E.; Alves, D.; Cachulo, M.L.; Khawaja, A.P.; Cougnard-Gregoire, A.; Merle, B.M.J.; Korb, C.; Erke, M.G.; et al. Prevalence of Age-Related Macular Degeneration in Europe: The Past and the Future. Ophthalmology 2017, 124, 1753–1763.
- Zhou, H.; Zhang, H.; Yu, A.; Xie, J. Association between sunlight exposure and risk of age-related macular degeneration: A meta-analysis. BMC Ophthalmol. 2018, 18, 331.
- Kuan, V.; Warwick, A.; Hingorani, A.; Tufail, A.; Cipriani, V.; Burgess, S.; Sofat, R.; International, A.M.D.G.C. Association of Smoking, Alcohol Consumption, Blood Pressure, Body Mass Index, and Glycemic Risk Factors With Age-Related Macular Degeneration: A Mendelian Randomization Study. JAMA Ophthalmol. 2021, 139, 1299–1306.
- Vujosevic, S.; Alovisi, C.; Chakravarthy, U. Epidemiology of geographic atrophy and its precursor features of intermediate age-related macular degeneration. Acta Ophthalmol. 2023, 101, 839–856.
- Ranganathan, S.; Male, D.A.; Ormsby, R.J.; Giannakis, E.; Gordon, D.L. Pinpointing the putative heparin/sialic acid-binding residues in the ‘sushi’ domain 7 of factor H: A molecular modeling study. Pac. Symp. Biocomput. 2000, 2000, 155–167.
- Despriet, D.D.; Klaver, C.C.; Witteman, J.C.; Bergen, A.A.; Kardys, I.; de Maat, M.P.; Boekhoorn, S.S.; Vingerling, J.R.; Hofman, A.; Oostra, B.A.; et al. Complement factor H polymorphism, complement activators, and risk of age-related macular degeneration. JAMA 2006, 296, 301–309.
- Edwards, A.O.; Ritter, R., 3rd; Abel, K.J.; Manning, A.; Panhuysen, C.; Farrer, L.A. Complement factor H polymorphism and age-related macular degeneration. Science 2005, 308, 421–424.
- Ram, S.; Sharma, A.K.; Simpson, S.D.; Gulati, S.; McQuillen, D.P.; Pangburn, M.K.; Rice, P.A. A novel sialic acid binding site on factor H mediates serum resistance of sialylated Neisseria gonorrhoeae. J. Exp. Med. 1998, 187, 743–752.
- Meri, S. Self-nonself discrimination by the complement system. FEBS Lett. 2016, 590, 2418–2434.
- Giannakis, E.; Male, D.A.; Ormsby, R.J.; Mold, C.; Jokiranta, T.S.; Ranganathan, S.; Gordon, D.L. Multiple ligand binding sites on domain seven of human complement factor H. Int. Immunopharmacol. 2001, 1, 433–443.
- Kraus, D.; Medof, M.E.; Mold, C. Complementary recognition of alternative pathway activators by decay-accelerating factor and factor H. Infect. Immun. 1998, 66, 399–405.
- Scholl, H.P.; Fleckenstein, M.; Charbel Issa, P.; Keilhauer, C.; Holz, F.G.; Weber, B.H. An update on the genetics of age-related macular degeneration. Mol. Vis. 2007, 13, 196–205.
- Micklisch, S.; Lin, Y.; Jacob, S.; Karlstetter, M.; Dannhausen, K.; Dasari, P.; von der Heide, M.; Dahse, H.M.; Schmolz, L.; Grassmann, F.; et al. Age-related macular degeneration associated polymorphism rs10490924 in ARMS2 results in deficiency of a complement activator. J. Neuroinflammation 2017, 14, 4.
- Munoz, S.S.; Li, H.; Ruberu, K.; Chu, Q.; Saghatelian, A.; Ooi, L.; Garner, B. The serine protease HtrA1 contributes to the formation of an extracellular 25-kDa apolipoprotein E fragment that stimulates neuritogenesis. J. Biol. Chem. 2018, 293, 4071–4084.
- Windham, I.A.; Cohen, S. The cell biology of APOE in the brain. Trends Cell Biol. 2023. ahead of print.
- Meri, S.; Haapasalo, K. Function and Dysfunction of Complement Factor H During Formation of Lipid-Rich Deposits. Front. Immunol. 2020, 11, 611830.
- Murray, I.J.; Rodrigo-Diaz, E.; Kelly, J.M.F.; Aslam, T.M.; Tahir, H.J.; Carden, D.; Patryas, L.; Parry, N.R.A. The role of dark adaptation in understanding early AMD. Prog. Retin. Eye Res. 2022, 88, 101015.
- Tolentino, M.J.; Miller, S.; Gaudio, A.R.; Sandberg, M.A. Visual field deficits in early age-related macular degeneration. Vis. Res. 1994, 34, 409–413.
- Remulla, J.F.; Gaudio, A.R.; Miller, S.; Sandberg, M.A. Foveal electroretinograms and choroidal perfusion characteristics in fellow eyes of patients with unilateral neovascular age-related macular degeneration. Br. J. Ophthalmol. 1995, 79, 558–561.
- Sandberg, M.A.; Gaudio, A.R. Slow photostress recovery and disease severity in age-related macular degeneration. Retina 1995, 15, 407–412.
- Owsley, C.; Huisingh, C.; Clark, M.E.; Jackson, G.R.; McGwin, G., Jr. Comparison of Visual Function in Older Eyes in the Earliest Stages of Age-related Macular Degeneration to Those in Normal Macular Health. Curr. Eye Res. 2016, 41, 266–272.
- Owsley, C.; McGwin, G., Jr.; Clark, M.E.; Jackson, G.R.; Callahan, M.A.; Kline, L.B.; Witherspoon, C.D.; Curcio, C.A. Delayed Rod-Mediated Dark Adaptation Is a Functional Biomarker for Incident Early Age-Related Macular Degeneration. Ophthalmology 2016, 123, 344–351.
- Panorgias, A.; Zawadzki, R.J.; Capps, A.G.; Hunter, A.A.; Morse, L.S.; Werner, J.S. Multimodal assessment of microscopic morphology and retinal function in patients with geographic atrophy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 4372–4384.
- Evans, J.R.; Lawrenson, J.G. Antioxidant vitamin and mineral supplements for slowing the progression of age-related macular degeneration. Cochrane Database Syst. Rev. 2023, 9, CD000254.
- Chew, E.Y.; Clemons, T.E.; Agron, E.; Domalpally, A.; Keenan, T.D.L.; Vitale, S.; Weber, C.; Smith, D.C.; Christen, W.; Group, A.R. Long-term Outcomes of Adding Lutein/Zeaxanthin and omega-3 Fatty Acids to the AREDS Supplements on Age-Related Macular Degeneration Progression: AREDS2 Report 28. JAMA Ophthalmol. 2022, 140, 692–698.
- Seddon, J.M.; Gensler, G.; Milton, R.C.; Klein, M.L.; Rifai, N. Association between C-reactive protein and age-related macular degeneration. JAMA 2004, 291, 704–710.
- Agron, E.; Mares, J.; Clemons, T.E.; Swaroop, A.; Chew, E.Y.; Keenan, T.D.L.; AREDS and AREDS2 Research Groups. Dietary Nutrient Intake and Progression to Late Age-Related Macular Degeneration in the Age-Related Eye Disease Studies 1 and 2. Ophthalmology 2021, 128, 425–442.
- Galuszka, M.; Pojda-Wilczek, D.; Karska-Basta, I. Age-Related Macular or Retinal Degeneration? Medicina 2023, 59, 920.
- Walter, P.; Widder, R.A.; Luke, C.; Konigsfeld, P.; Brunner, R. Electrophysiological abnormalities in age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 1999, 237, 962–968.
- Bartz-Schmidt, K.U.; Brunner, R.; Esser, P.; Luke, C.; Walter, P.; Sickel, W. The triple flash electroretinogram and its significance in macular diseases. B-wave recovery as a diagnostic tool. Graefes Arch. Clin. Exp. Ophthalmol. 1996, 234, 604–611.
- Gonzalez-Garcia, E.; Vilela, C.; Navea, A.; Arnal, E.; Muriach, M.; Romero, F.J. Electrophysiological and clinical tests in dry age-related macular degeneration follow-up: Differences between mfERG and OCT. Doc. Ophthalmol. 2016, 133, 31–39.
- Forrester, J.V. Bowman lecture on the role of inflammation in degenerative disease of the eye. Eye 2013, 27, 340–352.
- Ardeljan, D.; Chan, C.C. Aging is not a disease: Distinguishing age-related macular degeneration from aging. Prog. Retin. Eye Res. 2013, 37, 68–89.
- Chen, M.; Luo, C.; Zhao, J.; Devarajan, G.; Xu, H. Immune regulation in the aging retina. Prog. Retin. Eye Res. 2019, 69, 159–172.
- Chen, M.; Xu, H. Parainflammation, chronic inflammation, and age-related macular degeneration. J. Leukoc. Biol. 2015, 98, 713–725.
- Zhang, Y.; Wong, W.T. Innate Immunity in Age-Related Macular Degeneration. Adv. Exp. Med. Biol. 2021, 1256, 121–141.
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435.
- Chen, M.; Forrester, J.V.; Xu, H. Dysregulation in retinal para-inflammation and age-related retinal degeneration in CCL2 or CCR2 deficient mice. PLoS ONE 2011, 6, e22818.
- Goto, M. Inflammaging (inflammation + aging): A driving force for human aging based on an evolutionarily antagonistic pleiotropy theory? Biosci. Trends 2008, 2, 218–230.
- Tolentino, M.J.; Tolentino, A.J. Investigational drugs in clinical trials for macular degeneration. Expert Opin. Investig. Drugs 2022, 31, 1067–1085.
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218.
- Fletcher, A.E.; Bentham, G.C.; Agnew, M.; Young, I.S.; Augood, C.; Chakravarthy, U.; de Jong, P.T.; Rahu, M.; Seland, J.; Soubrane, G.; et al. Sunlight exposure, antioxidants, and age-related macular degeneration. Arch. Ophthalmol. 2008, 126, 1396–1403.
- Miralles de Imperial-Ollero, J.A.; Gallego-Ortega, A.; Ortin-Martinez, A.; Villegas-Perez, M.P.; Valiente-Soriano, F.J.; Vidal-Sanz, M. Animal Models of LED-Induced Phototoxicity. Short- and Long-Term In Vivo and Ex Vivo Retinal Alterations. Life 2021, 11, 1137.
- Busch, C.J.; Binder, C.J. Malondialdehyde epitopes as mediators of sterile inflammation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 398–406.
- Ye, F.; Kaneko, H.; Hayashi, Y.; Takayama, K.; Hwang, S.J.; Nishizawa, Y.; Kimoto, R.; Nagasaka, Y.; Tsunekawa, T.; Matsuura, T.; et al. Malondialdehyde induces autophagy dysfunction and VEGF secretion in the retinal pigment epithelium in age-related macular degeneration. Free Radic. Biol. Med. 2016, 94, 121–134.
- Hollyfield, J.G.; Bonilha, V.L.; Rayborn, M.E.; Yang, X.; Shadrach, K.G.; Lu, L.; Ufret, R.L.; Salomon, R.G.; Perez, V.L. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat. Med. 2008, 14, 194–198.
- Hollyfield, J.G.; Perez, V.L.; Salomon, R.G. A hapten generated from an oxidation fragment of docosahexaenoic acid is sufficient to initiate age-related macular degeneration. Mol. Neurobiol. 2010, 41, 290–298.
- Suzuki, M.; Tsujikawa, M.; Itabe, H.; Du, Z.J.; Xie, P.; Matsumura, N.; Fu, X.; Zhang, R.; Sonoda, K.H.; Egashira, K.; et al. Chronic photo-oxidative stress and subsequent MCP-1 activation as causative factors for age-related macular degeneration. J. Cell Sci. 2012, 125, 2407–2415.
- Zhang, M.; Xu, G.; Liu, W.; Ni, Y.; Zhou, W. Role of fractalkine/CX3CR1 interaction in light-induced photoreceptor degeneration through regulating retinal microglial activation and migration. PLoS ONE 2012, 7, e35446.
- Bearelly, S.; Khanifar, A.A.; Lederer, D.E.; Lee, J.J.; Ghodasra, J.H.; Stinnett, S.S.; Cousins, S.W. Use of fundus autofluorescence images to predict geographic atrophy progression. Retina 2011, 31, 81–86.
- Su, N.; Hansen, U.; Plagemann, T.; Gaher, K.; Leclaire, M.D.; Konig, J.; Hohn, A.; Grune, T.; Uhlig, C.E.; Eter, N.; et al. Sub-Retinal Injection of Human Lipofuscin in the Mouse—A Model of “Dry” Age-Related Macular Degeneration? Aging Dis. 2023, 14, 184–203.
- Ben-Shabat, S.; Itagaki, Y.; Jockusch, S.; Sparrow, J.R.; Turro, N.J.; Nakanishi, K. Formation of a nonaoxirane from A2E, a lipofuscin fluorophore related to macular degeneration, and evidence of singlet oxygen involvement. Angew. Chem. Int. Ed. Engl. 2002, 41, 814–817.
- Jager, R.D.; Mieler, W.F.; Miller, J.W. Age-related macular degeneration. N. Engl. J. Med. 2008, 358, 2606–2617.
- Nita, M.G.A. Antioxidative Role of Heterophagy, Autophagy, and Mitophagy in the Retina and Their Association with the Age-Related Macular Degeneration (AMD) Etiopathogenesis. Antioxidant 2023, 12, 1368.
- Kaarniranta, K.; Xu, H.; Kauppinen, A. Mechanistical retinal drug targets and challenges. Adv. Drug Deliv. Rev. 2018, 126, 177–184.
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431.
- Golestaneh, N.; Chu, Y.; Xiao, Y.Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2017, 8, e2537.
- Kaarniranta, K.; Sinha, D.; Blasiak, J.; Kauppinen, A.; Vereb, Z.; Salminen, A.; Boulton, M.E.; Petrovski, G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013, 9, 973–984.
- Osburn, W.O.; Yates, M.S.; Dolan, P.D.; Chen, S.; Liby, K.T.; Sporn, M.B.; Taguchi, K.; Yamamoto, M.; Kensler, T.W. Genetic or pharmacologic amplification of nrf2 signaling inhibits acute inflammatory liver injury in mice. Toxicol. Sci. 2008, 104, 218–227.
- Dinkova-Kostova, A.T.; Baird, L.; Holmstrom, K.M.; Meyer, C.J.; Abramov, A.Y. The spatiotemporal regulation of the Keap1-Nrf2 pathway and its importance in cellular bioenergetics. Biochem. Soc. Trans. 2015, 43, 602–610.
- Sachdeva, M.M.; Cano, M.; Handa, J.T. Nrf2 signaling is impaired in the aging RPE given an oxidative insult. Exp. Eye Res. 2014, 119, 111–114.
- Kamei, M.; Yoneda, K.; Kume, N.; Suzuki, M.; Itabe, H.; Matsuda, K.; Shimaoka, T.; Minami, M.; Yonehara, S.; Kita, T.; et al. Scavenger receptors for oxidized lipoprotein in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1801–1807.
- Park, D.H.; Connor, K.M.; Lambris, J.D. The Challenges and Promise of Complement Therapeutics for Ocular Diseases. Front. Immunol. 2019, 10, 1007.
- Spaide, R.F.; Vavvas, D.G. Complement Inhibition for Geographic Atrophy: Review of Salient Functional Outcomes and Perspective. Retina 2023, 43, 1064–1069.
- Halawa, O.A.; Lin, J.B.; Miller, J.W.; Vavvas, D.G. A Review of Completed and Ongoing Complement Inhibitor Trials for Geographic Atrophy Secondary to Age-Related Macular Degeneration. J. Clin. Med. 2021, 10, 2580.
- Lin, J.B.; Halawa, O.A.; Miller, J.W.; Vavvas, D.G. Complement Inhibition for Geographic Atrophy: A Tempting Target with Mixed Results. J. Clin. Med. 2021, 10, 2890.
- Johnson, L.V.; Leitner, W.P.; Staples, M.K.; Anderson, D.H. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp. Eye Res. 2001, 73, 887–896.
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 14682–14687.
- Mullins, R.F.; Russell, S.R.; Anderson, D.H.; Hageman, G.S. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000, 14, 835–846.
- Lin, J.B.; Serghiou, S.; Miller, J.W.; Vavvas, D.G. Systemic Complement Activation Profiles in Nonexudative Age-Related Macular Degeneration: A Meta-Analysis. J. Clin. Med. 2022, 11, 2371.
- Heier, J.S.; Lad, E.M.; Holz, F.G.; Rosenfeld, P.J.; Guymer, R.H.; Boyer, D.; Grossi, F.; Baumal, C.R.; Korobelnik, J.F.; Slakter, J.S.; et al. Pegcetacoplan for the treatment of geographic atrophy secondary to age-related macular degeneration (OAKS and DERBY): Two multicentre, randomised, double-masked, sham-controlled, phase 3 trials. Lancet 2023, 402, 1434–1448.
- Khanani, A.M.; Patel, S.S.; Staurenghi, G.; Tadayoni, R.; Danzig, C.J.; Eichenbaum, D.A.; Hsu, J.; Wykoff, C.C.; Heier, J.S.; Lally, D.R.; et al. Efficacy and safety of avacincaptad pegol in patients with geographic atrophy (GATHER2): 12-month results from a randomised, double-masked, phase 3 trial. Lancet 2023, 402, 1449–1458.
- Ho, J.; Witkin, A.J.; Liu, J.; Chen, Y.; Fujimoto, J.G.; Schuman, J.S.; Duker, J.S. Documentation of intraretinal retinal pigment epithelium migration via high-speed ultrahigh-resolution optical coherence tomography. Ophthalmology 2011, 118, 687–693.
- Wang, S.; Wang, X.; Cheng, Y.; Ouyang, W.; Sang, X.; Liu, J.; Su, Y.; Liu, Y.; Li, C.; Yang, L.; et al. Autophagy Dysfunction, Cellular Senescence, and Abnormal Immune-Inflammatory Responses in AMD: From Mechanisms to Therapeutic Potential. Oxid. Med. Cell Longev. 2019, 2019, 3632169.
- Xu, H.; Chen, M.; Forrester, J.V. Para-inflammation in the aging retina. Prog. Retin. Eye Res. 2009, 28, 348–368.
- Langmann, T. Microglia activation in retinal degeneration. J. Leukoc. Biol. 2007, 81, 1345–1351.
- Damani, M.R.; Zhao, L.; Fontainhas, A.M.; Amaral, J.; Fariss, R.N.; Wong, W.T. Age-related alterations in the dynamic behavior of microglia. Aging Cell 2011, 10, 263–276.
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318.
- Coscas, G.; De Benedetto, U.; Coscas, F.; Li Calzi, C.I.; Vismara, S.; Roudot-Thoraval, F.; Bandello, F.; Souied, E. Hyperreflective dots: A new spectral-domain optical coherence tomography entity for follow-up and prognosis in exudative age-related macular degeneration. Ophthalmologica 2013, 229, 32–37.
- Vujosevic, S.; Bini, S.; Midena, G.; Berton, M.; Pilotto, E.; Midena, E. Hyperreflective intraretinal spots in diabetics without and with nonproliferative diabetic retinopathy: An in vivo study using spectral domain OCT. J. Diabetes Res. 2013, 2013, 491835.
- Killingsworth, M.C.; Sarks, J.P.; Sarks, S.H. Macrophages related to Bruch’s membrane in age-related macular degeneration. Eye 1990, 4 Pt 4, 613–621.
- Penfold, P.L.; Killingsworth, M.C.; Sarks, S.H. Senile macular degeneration: The involvement of immunocompetent cells. Graefes Arch. Clin. Exp. Ophthalmol. 1985, 223, 69–76.
- Cherepanoff, S.; McMenamin, P.; Gillies, M.C.; Kettle, E.; Sarks, S.H. Bruch’s membrane and choroidal macrophages in early and advanced age-related macular degeneration. Br. J. Ophthalmol. 2010, 94, 918–925.
- Karlen, S.J.; Miller, E.B.; Wang, X.; Levine, E.S.; Zawadzki, R.J.; Burns, M.E. Monocyte infiltration rather than microglia proliferation dominates the early immune response to rapid photoreceptor degeneration. J. Neuroinflammation 2018, 15, 344.
- Klaus, C.; Liao, H.; Allendorf, D.H.; Brown, G.C.; Neumann, H. Sialylation acts as a checkpoint for innate immune responses in the central nervous system. Glia 2021, 69, 1619–1636.
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438.
- Ransohoff, R.M.; El Khoury, J. Microglia in Health and Disease. Cold. Spring Harb. Perspect. Biol. 2015, 8, a020560.
- Roche, S.L.; Wyse-Jackson, A.C.; Ruiz-Lopez, A.M.; Byrne, A.M.; Cotter, T.G. Fractalkine-CX3CR1 signaling is critical for progesterone-mediated neuroprotection in the retina. Sci. Rep. 2017, 7, 43067.
- Marneros, A.G. Role of inflammasome activation in neovascular age-related macular degeneration. FEBS J. 2023, 290, 28–36.
- Dieckmann, B.W.; Paguaga, M.E.; McCollum, G.W.; Penn, J.S.; Uddin, I. Role of NLRP3 Inflammasomes in Monocyte and Microglial Recruitments in Choroidal Neovascularization; Research Square: Durham, NC, USA, 2023.
- Mettu, P.S.; Allingham, M.J.; Cousins, S.W. Incomplete response to Anti-VEGF therapy in neovascular AMD: Exploring disease mechanisms and therapeutic opportunities. Prog. Retin. Eye Res. 2021, 82, 100906.
- Yang, Y.; Liu, F.; Tang, M.; Yuan, M.; Hu, A.; Zhan, Z.; Li, Z.; Li, J.; Ding, X.; Lu, L. Macrophage polarization in experimental and clinical choroidal neovascularization. Sci. Rep. 2016, 6, 30933.
- Cruz-Pimentel, M.; Wu, L. Complement Inhibitors for Advanced Dry Age-Related Macular Degeneration (Geographic Atrophy): Some Light at the End of the Tunnel? J. Clin. Med. 2023, 12, 5131.
- Bonilha, V.L.; Bell, B.A.; Hu, J.; Milliner, C.; Pauer, G.J.; Hagstrom, S.A.; Radu, R.A.; Hollyfield, J.G. Geographic Atrophy: Confocal Scanning Laser Ophthalmoscopy, Histology, and Inflammation in the Region of Expanding Lesions. Investig. Ophthalmol. Vis. Sci. 2020, 61, 15.
- Penfold, P.L.; Killingsworth, M.C.; Sarks, S.H. Senile macular degeneration. The involvement of giant cells in atrophy of the retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 1986, 27, 364–371.
- Sarks, S.H. Ageing and degeneration in the macular region: A clinico-pathological study. Br. J. Ophthalmol. 1976, 60, 324–341.
- Sarks, S.H. Drusen patterns predisposing to geographic atrophy of the retinal pigment epithelium. Aust. J. Ophthalmol. 1982, 10, 91–97.
- Koncz, G.; Jenei, V.; Toth, M.; Varadi, E.; Kardos, B.; Bacsi, A.; Mazlo, A. Damage-mediated macrophage polarization in sterile inflammation. Front. Immunol. 2023, 14, 1169560.
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483.
- Wang, Y.; Xiong, X.; Wang, K.; Bao, Y.; Zhang, T.; Ainiwaer, D.; Wang, G.; Li, H.; Sun, Z. Peripheral Klotho protects the kidney and brain by regulating M2a/M2c macrophage polarization in d-gal-treated aged mice. Tissue Cell 2023, 82, 102049.
- Sapudom, J.; Karaman, S.; Mohamed, W.K.E.; Garcia-Sabate, A.; Quartey, B.C.; Teo, J.C.M. 3D in vitro M2 macrophage model to mimic modulation of tissue repair. NPJ Regen. Med. 2021, 6, 83.
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 2009, 29, 13435–13444.
- Kisucka, A.; Bimbova, K.; Bacova, M.; Galik, J.; Lukacova, N. Activation of Neuroprotective Microglia and Astrocytes at the Lesion Site and in the Adjacent Segments Is Crucial for Spontaneous Locomotor Recovery after Spinal Cord Injury. Cells 2021, 10, 1943.
- Goerdt, S.; Politz, O.; Schledzewski, K.; Birk, R.; Gratchev, A.; Guillot, P.; Hakiy, N.; Klemke, C.D.; Dippel, E.; Kodelja, V.; et al. Alternative versus classical activation of macrophages. Pathobiology 1999, 67, 222–226.
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468.
- Devanney, N.A.; Stewart, A.N.; Gensel, J.C. Microglia and macrophage metabolism in CNS injury and disease: The role of immunometabolism in neurodegeneration and neurotrauma. Exp. Neurol. 2020, 329, 113310.
- Cao, X.; Shen, D.; Patel, M.M.; Tuo, J.; Johnson, T.M.; Olsen, T.W.; Chan, C.C. Macrophage polarization in the maculae of age-related macular degeneration: A pilot study. Pathol. Int. 2011, 61, 528–535.
- Ferrante, C.J.; Pinhal-Enfield, G.; Elson, G.; Cronstein, B.N.; Hasko, G.; Outram, S.; Leibovich, S.J. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Ralpha) signaling. Inflammation 2013, 36, 921–931.
- Emilsson, V.; Gudmundsson, E.F.; Jonmundsson, T.; Jonsson, B.G.; Twarog, M.; Gudmundsdottir, V.; Li, Z.; Finkel, N.; Poor, S.; Liu, X.; et al. A proteogenomic signature of age-related macular degeneration in blood. Nat. Commun. 2022, 13, 3401.
- Wang, Y.; Neumann, H. Alleviation of neurotoxicity by microglial human Siglec-11. J. Neurosci. 2010, 30, 3482–3488.
- Rajesh, C.; Radhakrishnan, P. The (Sialyl) Tn antigen: Contributions to immunosuppression in gastrointestinal cancers. Front. Oncol. 2022, 12, 1093496.
- Mullins, R.F.; Hageman, G.S. Human ocular drusen possess novel core domains with a distinct carbohydrate composition. J. Histochem. Cytochem. 1999, 47, 1533–1540.
- Smith, B.A.H.; Bertozzi, C.R. The clinical impact of glycobiology: Targeting selectins, Siglecs and mammalian glycans. Nat. Rev. Drug Discov. 2021, 20, 217–243.
- Duan, S.; Paulson, J.C. Siglecs as Immune Cell Checkpoints in Disease. Annu. Rev. Immunol. 2020, 38, 365–395.
- Shahraz, A.; Lin, Y.; Mbroh, J.; Winkler, J.; Liao, H.; Lackmann, M.; Bungartz, A.; Zipfel, P.F.; Skerka, C.; Neumann, H. Low molecular weight polysialic acid binds to properdin and reduces the activity of the alternative complement pathway. Sci. Rep. 2022, 12, 5818.
- Wang, S.K.; Cepko, C.L. Targeting Microglia to Treat Degenerative Eye Diseases. Front. Immunol. 2022, 13, 843558.
- Alves, I.; Fernandes, A.; Santos-Pereira, B.; Azevedo, C.M.; Pinho, S.S. Glycans as a key factor in self and nonself discrimination: Impact on the breach of immune tolerance. FEBS Lett. 2022, 596, 1485–1502.
- Liao, H.; Klaus, C.; Neumann, H. Control of Innate Immunity by Sialic Acids in the Nervous Tissue. Int. J. Mol. Sci. 2020, 21, 5494.
- Duarte Azevedo, M.; Sander, S.; Tenenbaum, L. GDNF, A Neuron-Derived Factor Upregulated in Glial Cells during Disease. J. Clin. Med. 2020, 9, 456.
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369.
- Krishnan, A.S.V.; Patel, D.; Lad, A.; Greene, M.K.; Smyth, P.; Gallaher, S.A.; Herron, U.M.; Scott, C.J.; Genead, M.; Tolentino, M. PolySialic acid-nanoparticles inhibit macrophage mediated inflammation through Siglec agonism: A potential treatment for age related macular degeneration. Front. Immunol. 2023, 14, 1237016.
- Bucan, I.; Skunca Herman, J.; Jeroncic Tomic, I.; Gornik, O.; Vatavuk, Z.; Bucan, K.; Lauc, G.; Polasek, O. N-Glycosylation Patterns across the Age-Related Macular Degeneration Spectrum. Molecules 2022, 27, 1774.
More