Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Sara Massironi and Version 2 by Rita Xu.
Eosinophilic esophagitis (EoE) is a chronic inflammatory disease characterized by eosinophilic infiltration of the esophagus. It arises from a complex interplay of genetic predisposition (susceptibility loci), environmental triggers (allergens and dietary antigens), and a dysregulated immune response, mainly mediated by type 2 T helper cell (Th2)-released cytokines, such as interleukin (IL)-4, IL-5, and IL-13.
- eosinophilic esophagitis
- pathogenesis
- soluble inflammatory mediators
1. Introduction
Eosinophilic esophagitis (EoE) is a chronic immune-mediated disease characterized by esophageal inflammation and dysfunction triggered by an abnormal Th2 inflammatory response [1][2][3][4][5][1,2,3,4,5]. The central pathogenesis of EoE revolves around a dysregulated immune response triggered by exposure to allergens, often derived from food [2][6][2,6]. This immune response leads to inflammation and tissue damage in the esophagus, resulting in the characteristic symptoms and histologic changes seen in EoE patients. Key elements of its central pathogenesis include allergen sensitization, Th2 immune response, epithelial barrier dysfunction, eosinophil infiltration, fibrosis, and remodeling [2][4][2,4]. In addition, genetic factors and environmental triggers play pivotal roles in the genesis of the disease.
Individuals with EoE develop sensitization to certain food antigens or environmental allergens [7]. This sensitization involves the activation of immune cells, particularly Th2 cells, which play a central role in driving allergic responses. Sensitized Th2 cells release cytokines, including interleukin (IL)-4, IL-5, and IL-13, which trigger an exaggerated immune response [5]. These cytokines promote eosinophil infiltration, mast cell activation, and tissue remodeling in the esophagus. Eosinophils release cytotoxic proteins and other inflammatory mediators that contribute to tissue damage and inflammation. In addition, the epithelial barrier of the esophagus is compromised in EoE, allowing allergens to penetrate the tissue more easily. Epithelial cells also exhibit altered gene expression, such as the downregulation of genes involved in differentiation and junctional proteins, resulting in the disruption of esophageal mucosal integrity.
Persistent inflammation and tissue damage trigger fibrosis and the remodeling of esophageal tissue. This leads to structural changes, such as the narrowing of the esophagus (strictures) and reduced esophageal compliance, mainly resulting in dysphagia [8].
Genetic susceptibility may play a role in the development of EoE, as certain genetic variations have been associated with an increased risk for the disease [9][10][9,10]. However, the exact genetic factors that contribute to susceptibility to EoE are still under investigation [10]. In addition to genetic predisposition, environmental factors such as diet and exposure to allergens or environmental allergens are also thought to influence the development and exacerbation of EoE. Certain foods, such as milk, wheat, and eggs, are often cited as triggers [11].
Understanding the central pathogenesis of EoE is critical to develop targeted therapeutic strategies that can reduce inflammation, restore esophageal function, and relieve symptoms [12]. However, the complexity of the disease is underscored by the involvement of multiple cellular and molecular pathways, making the development of effective treatments a multifaceted challenge.
Standard treatment therapies include dietary modification [11][13][11,13], esophageal dilation, and pharmacologic therapy [13][14][13,14]. Effective pharmacologic therapies include corticosteroids, rapidly evolving biological therapies, and proton-pump inhibitors (PPIs) [15]. A variety of novel therapeutic strategies have been developed targeting different cellular and soluble mediators that contribute to the complex mechanisms of the disease [16]. The pursuit of tailored approaches promises to improve patient outcomes and usher in a new era of precision medicine for people struggling with this complicated disease.
2. Pathogenesis of Eosinophilic Esophagitis (EoE)
The pathogenesis of EoE involves a multifaceted interplay between genetic predisposition and environmental triggers, leading to a dysregulated immune response.2.1. Genetic and Environmental Factors and Autoimmunity
EoE has a strong genetic component, as evidenced by various susceptibility loci identified through genetic studies. First-degree relatives of EoE patients have a 10 to 64 times higher risk of developing the condition compared with the general population [17][18][17,18], with an incidence among siblings of 2.4% vs. 5.5 per 10.000 in the general population [1][12][1,12]. Studies on twins reported an EoE frequency of 41% in monozygotic twins and 24% in dizygotic twins [18]. Moreover, certain genes, including TSLP, calpain-14 (CAPN14), Krüppel-like factor 13 (KLF13), and EMSY, have been associated with an increased risk of EoE [17][19][20][21][17,19,20,21]. Notably, the interplay between genes such as CAPN14 and specific environmental exposures highlights the complexity of EoE’s genetic–environmental interaction [1][22][1,22]. In particular, an association was found between the overexpression of CAPN14 in the early postnatal period of life (during breastfeeding and hospitalization in an intensive care unit) and exposure to certain environmental factors [12]. Overall, genetic risk factors are neither necessary nor sufficient to favor EoE, although they clearly modulate the individual lifetime risk of a given carrier. Environmental factors such as diet, cesarean births, antibiotic use, formula feeding [9], PPI use during childhood, cold climates, indoor pollutants, and housing [10] have been linked to an increased EoE risk. Conversely, owning a furry pet in childhood and Helicobacter pylori infection are associated with a lower risk of EoE [10][12][23][10,12,23]. The role of the gut microbiota in immune regulation is increasingly recognized [24]. Exploring therapies that modulate the composition of the microbiota to mitigate inflammation and allergen sensitization holds potential for the treatment of EoE [25][26][27][28][29][25,26,27,28,29]. Specifically, patients with active EoE showed an increase in Haemophilus and Aggregatibacter species and a decrease in Firmicutes [30], and early interactions between epithelial cells and the esophageal microbiota were able to modulate CXCL16 expression and recruit invariant natural killer T cells to the esophageal epithelium [31]. In a study in mice, administration of Lactococcus lactis NCC 2287 resulted in the histologic remission of EoE [32], although further research is needed in this area. The association with autoimmunity is increasingly being investigated, as 6% of patients with EoE have concomitant psoriasis, psoriatic arthritis, rheumatoid arthritis, or Hashimoto’s thyroiditis [33]. Indeed, antibodies against transmembrane desmoglein-3 (DSG3) and collagen XVII (NC16A) appeared to be increased in the sera of EoE patients [34].2.2. Soluble Inflammatory Mediators of EoE
After allergen-mediated epithelial injury in the esophageal mucosa, several damage-associated molecular patterns (DAMPs) are released, contributing to the onset of a type 2 inflammatory response, a hallmark of EoE. Notable DAMPs include thymic stromal lymphopoietin (TSLP), interleukin (IL)-33, and IL-25. Their selective inhibition has been shown to prevent the development of specific food allergies [35]. DAMPs were among the first patterns to be recognized as molecules able to activate group 2 innate lymphoid cells (ILC2s), early effectors of type 2 mucosal immunity. TSLP, an epithelial-derived cytokine expressed by several cell types, promotes Th2-type immune responses by influencing dendritic cells (DCs) [36]. Overexpression of TSLP is observed in the esophageal mucosa of EoE patients [37], and specific TSLP gene polymorphisms strongly correlate with the development of EoE in children [38]. TSLP signaling promotes dendritic cell activation, and the differentiation of naïve CD4 T cells into T helper 2 (Th2) cells and their proliferation by inducing IL-4 gene transcription, and supports immunoglobulin (Ig) E production [39][40][39,40]. A TSLP-triggered basophil-dependent mouse model of EoE-like disease was developed, and neutralization of TSLP ameliorated the disease [37]. IL-33, which is stored in the nuclei of various cells, including intestinal epithelial cells, is released upon exposure to environmental antigens [41][42][41,42]. Its binding to the ST2 receptor on immune cells, including eosinophils and mast cells, triggers a type 2 inflammatory response through the expression of costimulatory molecules such as CD86, major histocompatibility complex class II (MHC-II), and IL-6 [43][44][45][43,44,45]. IL-33 contributes to the expression of epithelial adhesion molecules, the release of cytokines and chemokines such as CXCL8 and CCL2, and eosinophil survival [46]. Increased levels of IL-33 have been described in the esophageal mucosa of patients with EoE [47], where it is expressed by both the endothelium and a subset of undifferentiated, nondividing esophageal epithelial cells in the basal layer [47][48][47,48]. Interestingly, a rare and novel chromosomal duplication of the entire IL-33 gene resulted in clinical features of EoE [49], whereas mouse models showed that the IL-33–ST2 axis is necessary to induce EoE [50] and to ensure homeostasis and the survival of eosinophils, as evidenced by reduced numbers of eosinophils in peripheral blood in IL-33- and ST2-deficient mice [51]. IL-25 belongs to the IL-17 cytokine family and is mainly expressed by Th2 cells and several epithelial cells. IL-25 activates immune cells and enhances IL-13 production by ILC2 in response to food allergens. It is produced in response to cell injury or tissue damage and activates immune cells through its interaction with the receptors IL-17RA and IL-17RB. IL-25 binding to the receptors activates several signaling pathways, such as NF-κB, MAPK, JAK, and STAT3, leading to several downstream effects such as inflammation and cell self-renewal, survival, or apoptosis [52]. Injection of exogenous IL-25 resulted in the proliferation of Th2 cells and ILC2 [53]. The effect of IL-25 on several allergy disorders has now been established [54]. As for EoE, the available data have shown that IL-25 is increased in active EoE compared with controls [55]. IL-4 and IL-13 play an important role in Th2 cell development. They are mainly expressed by basophils, eosinophils, mast cells, NK T cells, and ILC2 [56]. IL-4 and IL-13 share a common α-receptor, and they signal via the signal transducer and activator of transcription (STAT)-6, which allows them to exert specific functions in different cell types. They activate eosinophils and recruit them via CCL26 expression, regulate lymphocyte functions (Th2 differentiation and B cell IgG1 and IgE class switching) and macrophage M2 maturation, increase dendritic cell function [57], and promote epithelial barrier dysfunction by downregulating the epidermal differentiation complex (EDC) [58]. To date, IL-13 is considered the major effector cytokine in EoE, more abundant than IL-4 [57]. IL-13 actively induces the expression of VCAM-1/ICAM-1 in endothelial cells and IL-5 by lymphocytes, thereby promoting the migration of eosinophils from the bone marrow and inducing eosinophil homing to target organs [59]. IL-13 also acts on tissue remodeling by promoting epithelial cell hyperplasia, collagen deposition, and angiogenesis [60]. Lastly, its synergistic action with TGF-β1 can activate quiescent fibroblasts and differentiate them into myofibroblasts, and reduce the amplitude of esophageal muscle contraction [61]. Other inflammatory mediators such as IL-5, IL-18, IL-15, tumor necrosis factor alpha (TNF-α), the TNF-related cytokine LIGHT, and transforming growth factor β1 (TGF-β1) also contribute to EoE pathogenesis via different mechanisms. They modulate immune responses, eosinophil function, fibroblast activation, and tissue remodeling. IL-5 is expressed by Th2 lymphocytes, eosinophils, basophils, ILC2, CD34+ progenitor cells, and NKT cells. It induces the differentiation and maturation of eosinophils in bone marrow, homing in tissues, and protection from apoptosis [62][63][62,63]. Consequently, overexpression of IL-5 in patients with EoE is associated with disease activity and increased levels of eosinophils in the blood [64]. Of note, IL-5 also plays a role in basophil metabolism and function by causing them to increase histamine release upon activation [65]. IL-18 belongs to the IL-1 family, a pleiotropic cytokine produced mainly by macrophages, dendritic cells, and epithelial cells. Its overexpression has been noted in several atopic diseases, in which it exerts a pathological role by stimulating mast cell and basophil degranulation, recruiting granulocytes to the site of inflammation, inducing IgE production and isotype switching, and promoting a Th2 response [66][67][66,67]. It also induces invariant NK T cells to produce IL-5 and IL-13 [68]. Niranjanc et al. reported an increased concentration of IL-18 and its specific receptor IL-18Rα in the blood and esophageal mucosa of EoE patients, and this concentration correlated with the extent of esophageal eosinophilia, both in active and treated EoE patients [68]. IL-15 is predominantly produced by monocytes, macrophages, and dendritic cells. It stimulates the proliferation and differentiation of activated T cells and promotes the antigen-independent activation of NK cells [69]. IL-15 stimulates CD4+ T cells to produce the eosinophil-activating cytokines IL-5 and IL-13. It also induces esophageal epithelial cells to produce eosinophil-activating chemokines in mouse models and humans by expressing eotaxins-1–2 and eotaxin-3, respectively [70]. Tissue levels of IL-15 and IL-15Ra in the esophagus and blood levels of IL-15 were increased in patients with EoE compared with healthy individuals, and human IL-15 mRNA levels correlated with esophageal eosinophilia [64]. Tumor necrosis factor alpha (TNF-α) is an inflammatory cytokine with pleiotropic functions, among which it mediates the activation and survival of eosinophils. It is upregulated in EoE, particularly during active disease [71][72][71,72]. Upon esophageal inflammation due to external triggers, esophageal fibroblasts stimulate the release of TNF-α, which, in turn, promotes the epithelial-to-mesenchymal transition, fostering the development of esophageal fibrosis [73]. The TNF-related cytokine LIGHT is part of the TNF superfamily of cytokines that have emerged as important modulators of critical innate and adaptive immune responses. Recently, a major role of LIGHT in several eosinophilic disorders was described, including in EoE [74]. TGF-β1 is a pleiotropic cytokine involved in many different critical processes, including immune regulation, fibroblast activation, smooth muscle contraction, and the induction of the epithelial–mesenchymal transition. Patients with EoE have increased expression of TGF-β1 and its downstream nuclear transcription factor, phosphorylated SMAD2/3 protein [75]. Moreover, exposure to TGF-β1 correlates with a decreased expression of claudin-7, a tight junction membrane protein important for esophageal barrier function [76]. Consequently, chronic expression of TGF-β1 could allow allergen passage through the esophageal mucosa, leading to increased antigen presentation and immune activation. Nevertheless, there is conflicting evidence regarding the role of TGF-β1 in EoE, as some studies have shown that its mRNA levels are not increased in esophageal mucosa [77][78][77,78]. Eotaxins are a family of eosinophil-specific chemoattractant cytokines produced mainly by epithelial cells. They have been identified in several atopic diseases including EoE. They are mainly induced by IL-4 and IL-13, and they bind to the CCR3 receptor, which is predominantly expressed in eosinophils and mast cells [79], although they can also be released from activated eosinophils and mast cells. Notably, mice with a genetic deletion of CCR3 were protected from developing experimental EoE, demonstrating the role of eotaxins in the pathogenesis of the disease [71]. The eotaxin family includes eotaxins-1, -2, and -3, all of which are upregulated in EoE [80]. Among them, eotaxin-3 is the most abundant in EoE [71], and its expression is related to the concentrations of eosinophils and mast cells in esophageal biopsies [71]. Eotaxin-3 is encoded by the CCL26 gene, and the CCL26 concentration is able to distinguish EoE from healthy and GERD patients, respectively [80][81][80,81]. In contrast, the roles of eotaxins-1 and -2 in the pathogenesis of EoE appear to be marginal [72]. Indeed, deficiency in the former resulted in only modest attenuation of the disease [82][83][82,83], whereas the latter is mainly expressed in the lungs [84]. Finally, single-nucleotide polymorphisms in eotaxin-3 genes have been linked to EoE disease [85]. Although interferon-γ (IFNγ) is one of the major players in the type 1 inflammatory response, it has been reported to be increased in the mucosa of EoE patients [86]. In addition, an association with polymorphisms in a gene encoding a transcriptional regulator used by IFNγ has been described in patients with EoE [20]. Recently, in vitro treatment with IFNy was shown to increase esophageal barrier permeability and epithelial cell apoptosis [87], and EoE-causing allergens were able to stimulate CD4+ T cells to release IFNγ [86]. Overall, these data suggest a possible role of IFNγ in the pathogenesis of EoE, although further evidence is needed. Immunoglobulin G4 (IgG4) is an unusually dynamic antibody with unique molecular features distinct from other IgG subclasses. Biopsy specimens from adult and pediatric patients with EoE showed the deposition of higher levels of food-specific IgG4 antibodies [88][89][88,89]. Notably, the symptoms and histopathology of IgG4 deposition disappeared after the avoidance of cow milk and reappeared after its reintroduction [90]. The mechanism underlying the role of IgG4 in EoE disease needs further investigation. A concise overview of each inflammatory mediator’s function, mechanisms, and implications in eosinophilic esophagitis (EoE) is resumed in Figure 1.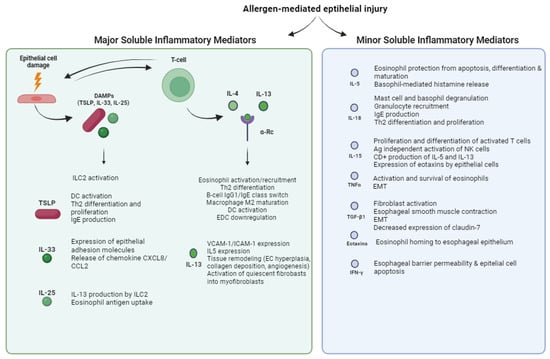
Figure 1. Functions of major and minor soluble inflammatory mediators of EoE. Allergen-mediated epithelial injury triggers the release of major and minor soluble inflammatory mediators, the former including damage-associated molecular patterns (DAMPs), IL-4, and IL-13, and the latter including IL-5, IL-18, IL-15, TNFα, TGF-β1, eotaxins, and IFNγ. TSLP = thymic stromal lymphopoietin; DC = dendritic cell; EC = epithelial cell; EMT = epithelial–mesenchymal transition; EDC = epidermal differentiation complex; α-Rc = α-Receptor.
2.3. Cellular Mediators of EoE
Eosinophils are the hallmark of the pathogenesis of EoE, and their importance has been studied in both mouse and human models. These leucocytes were originally thought to be purely destructive end-stage effector cells because of their ability to release toxic granule proteins. However, recent research has revealed a more complex role in EoE pathogenesis. The recruitment of eosinophils through the release of inflammatory signaling molecules, such as alarmins and eotaxins, represents the early physiological response to mucosal injury by environmental antigens to maintain local mucosal homeostasis. Chronic exposure to stimuli and tissue damage leads to the release of mediators such as GM-CSF and IL-5, which promote the activation and local recruitment of eosinophils [91]. Furthermore, the release of eosinophil peroxidase, eosinophil cationic protein, and major binding protein directly leads to tissue damage and the dysfunction of vagal muscarinic M2 receptors, resulting in esophageal dysmotility [92]. Finally, eosinophils might also act as antigen-presenting cells [93]. Despite the pivotal role of eosinophils in disease pathogenesis, mouse models genetically engineered to lack eosinophils have shown that clinical features such as esophageal motility disorder are independent of eosinophil inflammation [94], and randomized controlled trials of anti-IL-5 therapies have failed to achieve clinical remission ([95]; NCT04543409). Overall, the data suggest that EoE is not entirely dependent on eosinophils and that broader targeting of type 2 immunity may be required. Mastocytes are immune cells of the myeloid lineage traditionally associated with allergic reactions. They are classified according to whether they possess granules containing tryptase or tryptase and chymase, the latter being typical of esophageal mast cells both in diseases and under physiological conditions [96]. The trigger of mast cell activation in EoE is still unknown, as IgE-mediated mast-cell-dependent immediate responses to known food triggers have not yet been demonstrated, possibly due to the presence of non-IgE-mediated mechanisms [97]. Mast cells are major effectors in several atopic diseases, and their concentration and activation have been demonstrated in the esophageal mucosa of patients with EoE [94][98][99][100][101][94,98,99,100,101] and correlate with the extent of local infiltration of eosinophils [102] and EoE symptoms [96][103][96,103]. Mast cell degranulation, as detected by the presence of extracellular mast cell tryptase, was approximately 20-fold higher in EoE patients compared with control subjects [98]. Beyond their role in the type 2 inflammatory response, mast cells release specific mediators such as TGF-β1 tryptase, leukotrienes, prostaglandins, and histamine that contribute to esophageal smooth muscle hypertrophy and dysmotility [104][105][104,105]. Mast cells express the high-affinity receptor for IgE on their surfaces, which promotes a signal transduction process leading to the release of various inflammatory cytokines and chemokines [101]. Moreover, their ability to produce the eosinophil chemoattractant eotaxin-1 influences the accumulation of eosinophils in specific tissues [106], although some evidence for their ability to recruit eosinophils is still controversial [101]. Mast-cell-induced local inflammation of the esophagus results in altered mucosal permeability, smooth muscle hypertrophy, and altered contraction, leading to dysmotility [104][107][104,107]. Upon activation, mast cells promote tissue remodeling and fibrosis vis the expression of several profibrotic mediators such as TGF-β1 [108]. Decreased concentrations or altered functions of Tregs have been described in other atopic diseases [109][113], whereas their role in EoE pathogenesis is controversial, as their concentration has been reported to be either reduced [110][114] or increased [111][112][110,115] in mucosal samples. To date, none of the typical cytokines or interleukins associated with the Th2 response have been consistently and reproducibly altered to be used as a diagnostic tool for EoE. Compared with other atopic diseases (e.g., allergic asthma and atopic dermatitis), the lack of serological markers and unsatisfactory response to classic systemic immunosuppressive therapy in EoE may indeed reflect a different pathophysiology. According to recent theories, EoE may be a local, autonomous Th2 disease with a unique pathogenetic pathway that relies exclusively on components of the esophageal mucosa (i.e., TSLP) [3]. An important function of T cells in EoE is related to their expression of the TNF-related cytokine LIGHT, which can induce an inflammatory phenotype in fibroblasts [70]. The role of B cells in the pathogenesis of EoE has been poorly studied because of their minimal infiltration of the esophageal mucosa compared with other immune cells [72][113][72,109]. On the other hand, Vicario et Al. not only confirmed the increased density of B cells in EoE but also demonstrated that biopsy specimens from the esophageal mucosa expressed germline transcripts, underscoring the potential of B cells to undergo local class-switch recombination [114][116]. Under physiological conditions, the esophageal epithelium is relatively impermeable to medium- and large-size molecules, thus providing a barrier. In EoE, active inflammation results in damage to the epithelium associated with decreased expression of the structural proteins E-cadherin, desmoglein-1, involucrin, filaggrin, and synaptopodin [55][115][55,124], as well as the alteration of junctional proteins such as claudin and occludin [76][116][76,125]. The origin of these structural changes has been linked to the absence of the Kazal-type serine protease inhibitor (SPINK) 7, which is part of the differentiation program of the esophageal epithelium. SPINK7, which is highly expressed by healthy esophageal epithelial cells, was indeed significantly decreased in patients with EoE, who consequently exhibited higher permeability with dilated intercellular spaces [117][118][126,127]. Of note, the silencing of SPINK7 resulted in epithelial barrier dysfunction and activated transcriptional changes that stimulated the production of type 2 inflammatory responses [119][128]. Figure 2 summarizes the cellular mediators of EoE.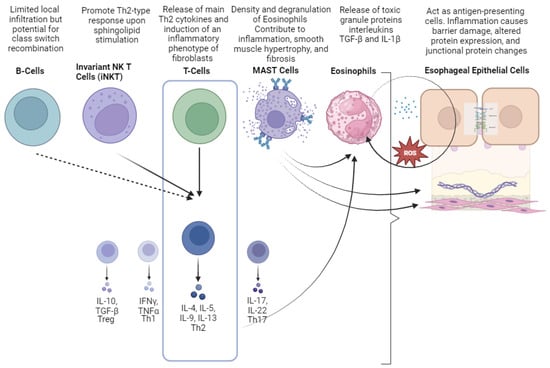
Figure 2. Cellular mediators involved in the pathogenesis of eosinophilic esophagitis (EoE). Different colors were used to represent and distinguish each cellular a soluble mediator.