Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Michael John Tolentino and Version 2 by Camila Xu.
Age-related macular degeneration (AMD) is an age-related condition that progressively impairs central vision with increasing age. AMD affects the central portion of the retina called the macula, which is required for central vision and visually demanding tasks like recognizing faces, reading, and driving.
- microglia
- macrophages
- macular degeneration
- sialic acid
- Siglecs
- nanoparticles
1. Age-Related Macular Degeneration (AMD)
1.1. Background
AMD is an age-related condition that progressively impairs central vision with increasing age. AMD affects the central portion of the retina called the macula, which is required for central vision and visually demanding tasks like recognizing faces, reading, and driving. Because central vision is important for these higher tasks, the impairment brought on by AMD results in decreased independence, mobility, and quality of life [1]. The potential for this form of vision loss is rated by surveyed individuals as one of the worst health outcomes possible [2].
AMD can be classified into early, intermediate, and late stages [3]. It is a disease that affects persons over the age of 60, and its incidence is increasing due to the rapid growth in the elderly population worldwide [4]. The prevalence of early- to intermediate-stage AMD in the US in 2019 reached approximately 18.34 million people [5]. This prevalence represents three times the number of patients with Alzheimer’s disease and is equal to all patients with a cancer diagnosis excluding melanoma [6][7][6,7]. By 2040, this disease that progressively causes central visual loss is estimated to affect 288 million people worldwide [8]. Currently, macular degeneration represents the third cause of vision loss secondary to ocular pathology worldwide [9]. Furthermore, the disease burden disproportionately affects less developed and low-income countries in Africa and Asia [10][11][10,11].
1.2. Clinical Presentation
The hallmark of early-stage AMD is the development of yellow subretinal deposits on the macula called drusen and/or abnormal pigmentary change called pigmentary mottling, clumping, or retinal pigment epithelial (RPE) change. When these changes worsen and reach a certain density, the stage is classified as intermediate. The late stage is reached when geographic atrophy (GA), neovascularization, or both develop [3].
From a visual function perspective, AMD can be divided into vision-threatening and non-vision-threatening stages. While drusen and RPE changes constitute the initiation of clinically detectable disease, these findings are usually not accompanied by any symptoms or noticeable vision loss [12]. Most patients with drusen and RPE changes alone do not develop significant vision loss. On the other hand, most patients progressing to the late-stage complications of geographic atrophy, exudative macular degeneration, or both will eventually develop moderate to severe central vision loss [13]. The prevalence of late-stage AMD in the US in 2019 was calculated to be 1.49 million people, which represents 7.5% of all the patients with AMD [5]. This difference between the prevalence of the non-vision-threatening early stage and vision-threatening late stage indicates that most patients will not develop moderate to severe central visual loss.
Macular degeneration is analogous to cardiovascular disease. Drusen and RPE changes are synonymous with plaque buildup on the coronary artery walls. As the drusen and RPE changes progress to intermediate and advanced stages, the risk of developing wet AMD or geographic atrophy increase [14]. In heart disease, advancing coronary artery plaque buildup results in coronary artery stenosis, which could lead to myocardial infarction, synonymous with wet macular degeneration, or ischemic cardiac heart failure, synonymous with geographic atrophy. Like cardiovascular disease, high cholesterol and coronary artery disease do not invariably lead to heart attack or cardiac heart failure.
Development of drusen and RPE changes, like plaque buildup and high cholesterol, are insidious and asymptomatic. These signs are predominantly detected by physician examination or sophisticated imaging tools such as optical coherence tomography (OCT) [15]. The initial development of exudative macular degeneration or geographic atrophy are often symptomatically undetectable, like ischemic heart disease or silent myocardial infarction, and require medical examination or sophisticated imaging technology to detect, such as wide-field ophthalmoscopy [16]. Exudative AMD often presents with mild visual symptoms such as metamorphopsia, distorted vision, color change, contrast abnormality, or mild visual loss [17]. In GA, the location of atrophy determines the severity of vision loss, where central-involving GA will result in the recognition of a blind spot while non-central-involving GA will be asymptomatic [18][19][18,19].
1.3. Risk Factors
Risk factors that have been consistently associated with AMD are age, ethnicity, smoking, and genetic polymorphisms. Age is the most important risk factor for the development and progression of both early- and late-stage AMD. This importance is demonstrated by the low 3.5% prevalence of early and 0.1% for late AMD in those younger than 59 and the high 17.5% prevalence of early and 9.8% for late AMD in those older than 85. This difference is a 5 (early) and 98 (late) times increase in prevalence between these two age groups [20]. In regard to ethnicity, white Europeans have the highest annual incidence of both early and late AMD [21]. Smoking is the strongest associated modifiable risk factor in both early- and late-stage AMD patients [22].
In total, 103 AMD genes have been associated with AMD, but the most significantly associated are the complement factor H (CFH), age-related maculopathy susceptibility 2/high-temperature requirement A serine peptidase 1 (ARMS2/HTRA1), and apolipoprotein E (APOE) polymorphisms [23]. The most associated polymorphism to AMD is found in the CFH loci: the substitution of the histidine for tyrosine at the 42 codon of chromosome 1-region 31 (rs1061170), which results in the alteration of the sialic acid/heparin binding site in the short consensus repeat region 7 of the CFH protein [24][25][26][24,25,26]. Effectively, this polymorphism reduces binding with self-associated molecular patterns (SAMPs) and permits unchecked alternative complement activation and resultant chronic immune activation [27][28][29][30][27,28,29,30].
ARMS2/HTRA1 are genes that exhibit a high degree of linkage disequilibrium, so it is difficult to determine which polymorphism is responsible for its association with AMD [31]. ARMS2 is a protein that is expressed by human monocytes, binds to apoptotic cells, and recruits properdin, which facilitates C3b opsonization and phagocytosis of apoptotic and necrotic cells [32]. The polymorphism in ARMS2 reduces phagocytosis of necrotic and apoptotic debris and may result in accumulation of drusen material. The absence of ARMS2 can result in both reduced phagocytosis of apoptotic cells and increased nonspecific phagocytosis of healthy cells, leading to cellular loss like geographic atrophy [32].
HTRA1 polymorphism results in increased production of the HTRA1 protein, which is a serine peptidase known to cleave APOE [33]. Increased cleavage would inactivate APOE, mimicking the dysfunction brought about by the polymorphisms in APOE found in AMD. APOE’s best-known role is to regulate the transportation of lipids and cholesterol in the retina and brain [34]. Another major role is to protect lipids from complement attack by binding and activating CFH and protecting high-density lipoproteins (HDLs) from complement attack [35]. Polymorphisms in APOE associated with AMD also reduce APOE levels, dysregulating lipid and cholesterol clearance and inciting inflammation by not protecting the lipids from complement attack [35].
2. Central Role of Inflammation and Parainflammation in AMD
2.1. Clinical Evidence of Inflammation
In the early and intermediate stages of macular degeneration, measurable photoreceptor dysfunction is reflected in abnormalities dark adaptation, visual field, photo stress, and electro-retinographic changes [36][37][38][39][36,37,38,39]. These disease-correlated changes in visual function demonstrate inflammation as the underlying pathology behind all stages of macular degeneration. Prolonged dark adaptation, which progressively worsens in step with stage of AMD, is caused by inflammatory visual cycle impairment and indicates a worsening of inflammation as AMD progresses [40][41][40,41]. In one study, patients with early AMD had qualitative visual changes and symptoms of distortion that could be detected and quantified by visual field analysis [37]. These deficits of form recognition and sensitivity were found in areas of RPE atrophy not defined as geographic atrophy within this patient population, indicating asymptomatic vision loss in areas of overactive inflammation [37]. Photo stress recovery time, a measure of visual pigment recycling time, was inversely correlated with visual acuity, and inflammation-induced prolongation of recovery time was directly correlated with the presence or absence of geographic atrophy and advancing age [39]. In late AMD, inflammatory slowing of implicit time and amplitude reduction on electroretinograms (ERGs) of patients with geographic atrophy were seen in areas bordering fundus autofluorescence-defined geographic atrophy [42]. Foveal ERG performed in fellow eyes of patients with wet AMD who did not have severe visual acuity changes demonstrated implicit time prolongation, indicating that fellow eyes were also undergoing inflammation [38]. Visual cycle alteration represented the first indication of the central role of inflammation in AMD.2.2. Anti-Oxidant Therapy for Early-Stage AMD
While clinical findings in AMD pointed to inflammation as the central driver of AMD progression, initial therapeutics for AMD were focused on anti-oxidation. To this day, the only consensus treatment for early/intermediate-stage AMD is the use of anti-oxidant therapies studied in a large National Eye Institute study called the Age-Related Eye Disease Study 1 and 2 (AREDS 1 and 2) [43]. This study has produced over 28 reports and countless publications on the benefit of anti-oxidant therapy for the prevention of late AMD progression [44]. While the conclusions from these studies demonstrated a reduction in the rate of development of large drusen, geographic atrophy, and exudative AMD, the modest reduction in rate of progression implicates oxidation as only a stimulator of inflammation rather than the main driver for AMD progression [45][46][45,46].2.3. Oxidation-Induced Dysfunctional Parainflammation in Early AMD
Demographic, environmental, and genomic risk factors combined with electrophysiological and psychophysical studies definitively implicate retinal inflammation as the major underlying factor in the development of all stages of AMD [47][48][49][50][47,48,49,50]. The degree of dysfunctional inflammation determines the stage and clinical presentation [51]. Multiple papers have described macular degeneration as a disease that is initiated by dysregulated parainflammation leading to the drusen stage of AMD then progressing to overt inflammation that triggers the late stage of AMD [52][53][54][55][52,53,54,55]. In 2008, Medzhitov postulated a parainflammatory state that lay between basal homeostatic conditions and true inflammation [56]. This parainflammatory state is considered an adaptive immune response to low level tissue stress such as the age-related accumulation of oxidative byproducts [57]. This acquired dysfunctional parainflammation that occurs with aging has been termed inflamaging and likely explains the early stages of AMD with drusen development and RPE changes [58]. The development of late exudative and geographic atrophy stages, on the other hand, is an overt innate immune activation with end-stage pathology determined by macrophage polarization [59]. Oxidative damage is the main tissue stress that stimulates parainflammation in the retina [60] (Figure 1(1)). The macula, which is exposed to photo, metabolic, phagocytic, and mitochondrial reactive oxygen species (ROS) production, is a site of tremendous oxidative stress. An association between blue light exposure (photo-oxidative light) and low anti-oxidant levels demonstrated an association with early and neovascular forms of AMD, implicating photo-oxidative light as a contributor to oxidative stress in AMD [61].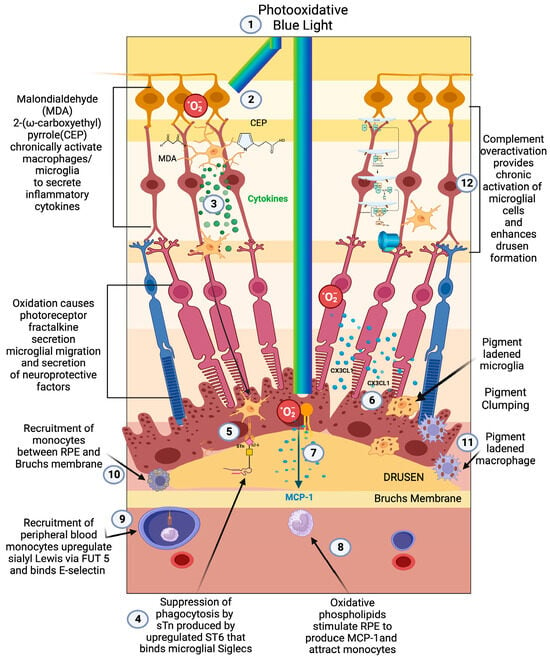
Figure 1. Early/intermediate-stage AMD. (1) Photo-oxidative blue light (2) oxidizes retinal lipids to produces oxidative byproducts (MDA, CEP) and reactive oxygen species, (3) which activate microglial cells to secrete cytokines. These activated macrophages are hindered from becoming phagocytic due to the (4) upregulation of ST6 that produces (5) sTN on the surface of retinal cells and drusen that agonizes Siglecs to prevent phagocytosis of drusen. (6) Reactive oxygen species also cause photoreceptors to secrete CX3CL1, which promotes migration of microglia and macrophages to the retina. (7) Oxidized phospholipids stimulate RPE cells to produce MCP—1 (8), which recruits peripheral blood monocytes (PBMCs) to areas of phospholipid oxidation. (9) Upregulation of FUT5 on PBMCs localizes monocytes to areas that are secreting chemokines such as CX3CL1 that upregulate e-selectin on vascular endothelium. (10) Monocytes are found between RPE cells and Bruch’s membrane. (11) Microglial cells and macrophages are also found to be pigment-ladened in this sub-RPE space and at the RPE cell layer, which appears as RPE pigment clumping. (12) The overactivation of complement caused by the polymorphism-induced impaired function of CFH will produce complement pathway metabolites that accumulate in drusen and can activate microglial cells.
2.4. Complement Pathway-Induced Parainflammation
Despite the strong association of complement factor polymorphisms with the development of AMD and the development of two FDA-approved treatments for geographic atrophy (Pegcetacoplan and Avacincaptad pegol), there is a consensus that complement does not fully account for disease development [82][83][84][85][82,83,84,85]. Its direct role in disease may be in the development of drusen, and it may play only an indirect role in the development of late-stage disease [86] (Figure 1(12)). Evidence to support this role in producing drusen is found in the proteomic analysis of drusen from retinas of patients with AMD, where a large proportion of patients had complement proteins 9 (C9) and 3(C3) in their drusen [87]. C9 is a protein found in conjunction with the c5-9 complex or membrane attack complex. This finding agreed with earlier histopathologic analysis that corroborated the presence of multiple complement factors in drusen [88]. A meta-analysis found that systemic complement overactivation was a feature associated with early/intermediate AMD rather than late-stage geographic atrophy [89]. While no trials have been performed to determine the effect of complement factor depletion on the development of drusen, several trials have looked at the effect of complement C3(Pegcetacoplan) and C5 (Avacincaptad pegol) depletion and demonstrated a modest reduction in the growth rate of GA [90][91][90,91]. According to both pre-clinical and clinical studies, complement overactivation is important in initiating AMD by exacerbating parainflammatory overactivation and dysfunction, which enhances drusen formation. The inability of profound complement depletion to halt the progression of late-stage GA relegates complement as a minor player in the pathogenesis of late-stage AMD [90][91][90,91].2.5. Early-Stage AMD Dysfunctional Parainflammation
At the cellular level, the early/intermediate stage is the accumulation of oxidized, metabolic, inflammatory debris that appears as yellow sub retinal deposits called drusen [87]. The pigmentary clumping seen in AMD represents pigment-ladened microglia or macrophage migration towards the retina, Bruch’s membrane, and under surface of the RPE [92] (Figure 1(9–11)). The accumulation of proinflammatory oxidative byproducts CEP and MDA leads to phagocytic/cytokine secreting microglial polarization, activation of parainflammatory mechanisms, and, potentially, recruitment of peripheral blood macrophages to the subretinal space to compensate for this accumulation of subretinal toxic substances [55] (Figure 1(2,3,10)). With age-dependent RPE cell senescence and dysfunction of autophagy, parainflammatory activation of microglial cells accelerates, resulting in the recruitment of peripheral blood-derived macrophages into the subretinal space [93].2.6. Microglia’s Central Role in Dysfunctional Parainflammation
Microglia play a central role in modulating parainflammation [94] (Figure 2(1)). Microglial overactivation represents a common pathomechanism in a variety of retinal degenerative diseases and is often overactivated prior to the onset of overt retinal cell death [95]. In the retina, microglia’s dynamic motility allows comprehensive surveillance coverage of the entire retina in a short time period [96] (Figure 2(5)). This motility allows microglia to interact with retinal neurons and macroglia and play a central role in retinal homeostatic maintenance and clearance of cellular and metabolic debris [97].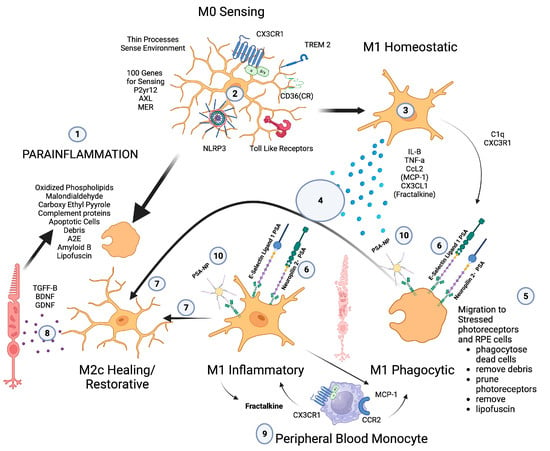
Figure 2. Microglial parainflammatory regulation: Microglia remove toxic metabolites, apoptotic cells, and oxidative debris while maintaining the health of photoreceptors and retinal cells in a process called parainflammation. (1) Parainflammation is initiated by pattern recognition receptors found on (2) M0 microglia such as TLR, TREM2, CX3CR1, NLRP3, and CD36, which use these receptors to bind and sense toxins, apoptotic cells, damage, and pathogen-associated molecular patterns. Once microglia are activated, they polarize to the (3) M1 like state, (4) secrete proinflammatory cytokines, and (5) migrate to the areas of stressed RPE cells and photoreceptors. (6) Upon activation, neuropilin-2-PSA and E-selectin ligand 1-PSA are secreted by these activated microglia. (6) The PSA on these glycoproteins binds Siglecs on activated microglial cells (7) to polarize them into the M2c healing state (8), which releases growth factors to protect and regenerate the stressed photoreceptors and RPE cells. (9) This homeostatic maintenance function of microglial parainflammation, if not adequately modulated with sialic acid checkpoint regulation, will result in recruitment of peripheral blood monocytes and AMD disease progression. (10) A PSA-nanoparticle (PSA-NP) mimics E-selectin ligand 1 PSA and Neuropilin 2-PSA to (7) polarize M1 activated microglia into the M2c healing state.
2.7. Peripheral Blood-Derived Macrophages’ Role in Transition to Late AMD
The major role of peripheral blood monocyte-derived macrophages in the progression of the early drusen stage to late-stage geographic atrophy and exudative AMD is evidenced by the increase in number of activated macrophages in the choroid and Bruch’s membrane as AMD progresses. The observation that the highest number of activated macrophages are found in eyes with choroidal neovascularization further supports this central role [102]. The recruitment of peripheral blood-derived macrophages is a major function of retinal microglial cells when injury or toxic material overwhelm the microglia’s ability to phagocytose toxic oxidative byproducts, injured cells, or apoptotic cells [103]. To maintain macular health, the microglia must phagocytose oxidative byproducts (oxidized phospholipids, malondialdehyde, carboxyethyl pyrrole), apoptotic cells, complement proteins, abnormal proteins (Amyloid B), and toxic metabolites (A2E). If microglia do not encounter appropriate checkpoint ligands in the form of sialic acid, then chemokine signaling will recruit peripheral blood macrophages [104] (Figure 2(9)). Patho-mechanistically, the recruitment of peripheral blood monocytes in clinically evident macular degeneration is mediated by microglial chemokine signaling, which is responsible for the recruitment of monocytes to the choroid and the retina. The chemokine receptors CCR2 and CX3CR1 and their respective ligands CCL2 (monocyte chemotactic protein-1, MCP-1) and CX3CL1 (fractalkine) mediate this recruitment [68][105][106][68,105,106]. While fractalkine-CX3CR1 signaling is proinflammatory, it is also critical for progesterone-mediated neuroprotection of the retina [107]. This dual role of Fractalkine-CX3CR1 interaction in microglial proinflammatory activation and neuroprotective properties demonstrates the necessity of tight regulation of microglial cells. If the proinflammatory properties of fractalkine-CX3CR1-activated microglia could be checked, but they could maintain their neuroprotective properties, then polarizing macrophages to the resolution state would eliminate inflammation and attenuate phagocytosis while providing neuroprotection. Without appropriate checkpoint regulation of microglial cells, the inflammatory activation state of microglia will promote more photoreceptor degeneration rather than rescue of photoreceptors.2.8. Macrophage Recruitment Indicator of Late-Stage AMD
The central role of macrophages in the pathogenesis of neovascular wet AMD is widely accepted [108][109][110][111][108,109,110,111], but the critical role of activated macrophages in the pathogenesis of geographic atrophy has been widely overlooked due to the focus on the complement pathway [112]. Prior to the genomic association between complement factors and AMD, histopathologic evidence pointed to macrophage/mononuclear phagocyte/multinucleated giant cells as the central causative factor in the pathogenesis of GA and the main phagocytic cause of retinal cellular clearance that manifests as RPE and photoreceptor loss [101][113][101,113] (Figure 3(5,6,9)).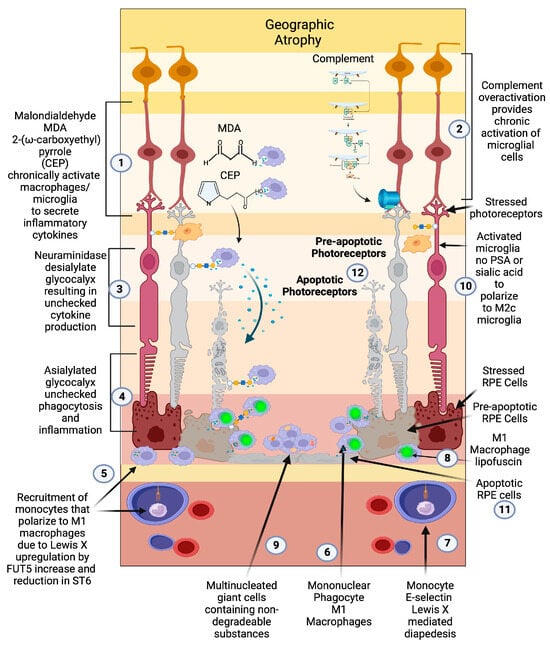
Figure 3. Geographic atrophy: (1) Progressive accumulation of oxidative byproducts (CEP, MDA) and (2) chronic overactivated complement pathway chronically activate microglia (3), which secrete neuraminidase and desialylate photoreceptors and RPE cells. (4) This loss of sialylation prevents restoration of homeostasis. (5) Chronically overactivated phagocytosis and inflammation recruits peripheral blood macrophages by upregulation of the fucosyltransferase FUT5 that produces Lewis X glycosylation on monocytes to bind E-selectin and (6) localize monocytes to sites of inflammation (7), allowing for diapedesis of the monocyte across the blood–retinal barrier. (8) These monocytes polarize to M1 macrophages when they enter the retina and are not able to clear substances like lipofuscin and other undegradable substances. (9) The macrophages form multinucleated giant cells because they phagocytose structures that are undegradable. (10) Since there is no sialic acid or polysialic acid to polarize to the healing M2c state, the macrophages are unchecked and result in elimination (11) first of the RPE cells then the (12) photoreceptors. The unchecked macrophages are the main determinant of growth of geographic atrophy.
2.9. Macrophage Polarization Determines Late-Stage AMD
The classically activated (M1) and alternatively activated (M2) binary description of macrophage polarization, based on biomarker expression and cytokine production, does not reflect the true character of these subtypes. A function-based description of macrophage polarization better characterizes their role in pathology [59][117][118][59,117,118]. The M1 polarization state is characterized as the proinflammatory phagocytic state. The M2 state can be subdivided into four M2 subtypes. The M2 a, b subtype are the anti-inflammatory pro-fibrotic type, the M2d is the pro angiogenic phenotype, and the M2c is the anti-inflammatory and neuroprotective type [117][118][119][117,118,119]. The M2c can also dedifferentiate myofibroblasts, so is considered anti-fibrotic [120]. In vitro, M1 macrophages are predominantly neurotoxic with modest axon growth-promoting effect, in contrast to the M2 macrophages, which promote long-distance axon growth without neurotoxicity [121]. In vivo studies in traumatic brain or spinal cord injury have characterized the time course and characteristics of M1/M2 polarization after injury [121][122][121,122]. M1-like macrophages release oxidative metabolites and proteases that kill neurons and glial cells [121]. In contrast, M2-like cells facilitate tissue repair [123]. In spinal cord injury models, increased M2c microglia expressed in the first week after injury correlated with better neurological outcome, indicating a healing neuroprotective function of M2c microglia/macrophages [122]. A time course study comparing M1 versus M2 levels in this model show that M1 expression is upregulated for at least a month, while M2 levels diminish drastically 1 week post injury. The level of M2a and M2c upregulation within the first week correlated well with neurological recovery. The neurological recovery resulted from the neuroprotective properties of microglia polarized to the M2c or M2a state. These properties were the secretion of trophic factors such as brain-derived neurotrophic factor (BDNF) and glial-derived neurotrophic factor (GDNF) and the ability to perform controlled phagocytosis, which prevents necrosis of surrounding tissue [124] (Figure 2(8)). While at first glance, macular degeneration may not appear to be a disease of acute CNS injury, the macrophage microglial behavior in the retina is consistent with what is seen in injury models of the CNS [55][125][55,125]. Eyes with advanced AMD had a higher ratio of M1 to M2 macrophages than age-matched normal autopsied eyes [126]. This enhanced chronic expression of neurotoxic phagocytic M1 macrophage with the reduction in M2c neuroprotective macrophages results in unchecked photoreceptor, RPE cell degeneration, and phagocytosis (geographic atrophy) [126]. The abundance of M1 polarized macrophages and adenosine polarize M1 to M2D VEGF-producing macrophages, resulting in the development of subretinal neovascularization [127]. If the M2 a, b polarization predominates, then retinal fibrosis will develop (disciform scar) [59]. Like CNS injury, the failure to polarize M1 macrophages towards the M2 c state will result in failure of functional recovery [122] (Figure 4).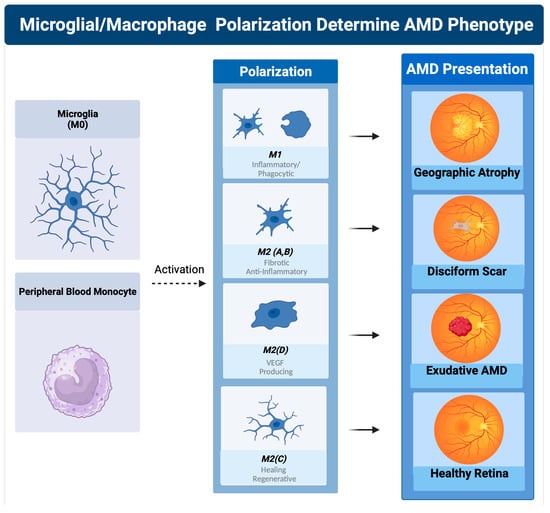
Figure 4. Macrophage polarization determines AMD phenotype. The plasticity and different polarization states correlate with the clinical picture seen in late-stage macular degeneration. In geographic atrophy, RPE cells and photoreceptors are phagocytosed as a function of the M1 polarized phagocytic macrophages. Exudative AMD is neovascularization produced by overexpression of VEGF, the main cytokine secreted by the M2d polarization state. The sequelae of exudative AMD untreated with anti-VEGF therapies is a disciform fibrotic scar in the control of the pro-fibrotic M2 A, B state. With appropriate sialic acid signaling such as PSA, all polarization states can transform into the healing M2c state.