Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Lara Baticic and Version 2 by Catherine Yang.
The postprandial state is known as the metabolic assessment period during and after a meal (6–12 h), which involves the digestion and absorption of nutrients, mainly fatty acids and carbohydrates from food. This state spans most of the day, more than 16 h, and is characterized by an increase in glycemia and lipidemia associated with systemic low-grade inflammation. Inflammation is an essential component of innate (nonspecific) immunity and host defense, but a chronic systemic low-grade inflammatory state is also the basis of the metabolic syndrome.
- cancer
- chronic low-grade inflammation
- diabetes
1. General Considerations
Diabetes mellitus (DM) is a chronic metabolic disease characterized by hyperglycemia resulting from defects in insulin secretion, insulin action or both. Chronic hyperglycemia due to a lack of insulin production or insulin resistance is associated with long-term damage, dysfunction and the failure of different organs and is a severe risk factor for various pathologies, such as cardiovascular, neurological and kidney diseases, among others [1][27]. DM is classified into two types, type 1 diabetes (T1D) and type 2 diabetes (T2D), the latter of which will be examined in this resviearch w. T2D is marked by insulin resistance and the productivity dysfunction of insulin secreted from β cells of the pancreas. These conditions are linked to smoking, aging, obesity and an incorrect lifestyle. In fact, patients suffering from T2D have high glucose levels and lipids in their blood which are indicative of a postprandial dysmetabolism condition [2][28].
Under physiological conditions, the release of insulin produced by β cells of the pancreas follows the glycemic curve during a meal with the purpose of reducing excessive plasma glucose levels, and it is accompanied by an oxidative and inflammatory response [3][29]. The insulin production in T2D patients does not grow proportionally to the glycemic load but is delayed, leading to higher and longer postprandial glucose excursions. Thus, the liver and peripheral tissues are not able to stimulate the uptake of glucose [4][30]. This condition also affects T2D patients’ lipid metabolism, as they have high levels of very-low-density lipoproteins (VLDLs), chylomicrons (CMs) and chylomicron remnants (CMRs) and low levels of high-density lipoproteins (HDLs) [5][31]. The increase in VLDLs is associated with the production of low-density LDLs, a subset of LDLs with a high triglyceride content that leads to an atherogenic risk. These conditions, along with insulin resistance, impair the uptake of fatty acids into muscle and lead to an increase in postprandial fatty acids in patients with T2D [6][7][32,33]. In the following sections, the features of T2D, the factors involved in this pathology, inflammation and its interactions with other organs are analyzed.
2. Insulin Resistance and Inflammation
The release of postprandial insulin, which is produced and released by β cells of the pancreas, is also controlled by incretins such as glucagon-like peptide 1 (GLP1) and glucose-dependent insulinotropic polypeptide (GIP) [8][34]. Normally, GLP1 and GIP are produced by the gastrointestinal tract during a meal, and their function consists of reducing the postprandial lipid concentration. T2D causes a low level of postprandial GLP1 together with an impairment of other incretin effects, leading to a high glucose peak after a meal [8][9][34,35].
As written above, insulin resistance is one of the main steps that lead to the development of T2D and it is associated with obesity, a lack of physical activity and aging; in T2D patients, in fact, insulin is not sufficient to act against the high level of glucose consumed during a meal [1][10][27,36]. Key players that were found to be involved in insulin resistance are inflammatory cytokines like TNFα, IL-6, C-reactive protein and plasminogen activator inhibitor, all of which were found to be elevated in obese mice [11][12][37,38]. These molecules, together with ROS and free fatty acids, activate Ikβα kinase β (IKKβ) and c-Jun N-terminal kinase 1 (JNK-1) which phosphorylate the serine residues of IRS protein and inhibit insulin signaling in liver and adipose tissue. In addition, IKKβ phosphorylates Ikβα and promotes its ubiquitination and degradation. During this process, it stimulates nuclear factor (NFkβ) translocation into the nucleus to activate the transcription of inflammatory genes [13][39].
The inhibition of insulin signaling may also impair the Glut-4 translocation to cell membranes, thus leading to hyperglycemia, through Janus kinase/signal transducers and the activator of transcription (JAK/STAT) pathway. As described by Ye J. et al., the perpetrator of JAK/STAT activation is JNK. JAK phosphorylates the tyrosine of STAT and then induces the dimerization and translocation of STAT to the nucleus that phosphorylates the serine residues on IRS-1 [14][40]. All these processes lead to hyperglycemia but not the translocation of GLUT-4 to cell membranes [13][39] and are schematically presented in Figure 15.
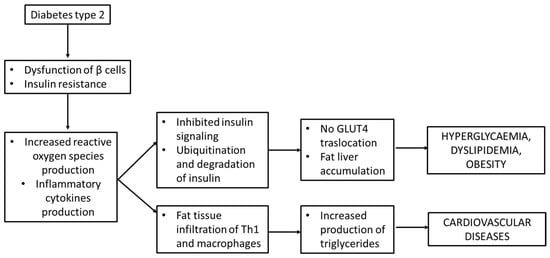
Figure 15.
Insulin resistance and inflammation in
diabetes mellitus
type 2.
3. Inflammation and Islets of Langerhans Failure
Pancreatic islets are composed of two compartments: the first one for the production of insulin and the second one made up of innate immune cells. Among the latter, macrophages do not follow the M2/M1 configuration like in adipose tissue, but they are found only in the M1 state, which occurs in healthy islets with a high expression of IL-1β, TNFα and the pro-inflammatory transcription factor interferon regulatory factor (IRF)-5 [15][16][41,42]. It seems that the function of macrophages is to monitor and enhance insulin secretion of β cells through endogenous ATP co-produced with insulin and the production of factors like retinoic acid [17][18][43,44].
The chronic state of obesity triggers negative conditions like hyperglycaemia and T2D, due to the insufficient production of insulin. This phenomenon is called “β cells failure” and consists of an inflammatory process, with the production of pro-inflammatory cytokines such as IL-1β, TNFα and chemokines ligand 2 (CCL-2). In addition, there is an increase in macrophages, which occurs in both humans affected with T2D and mice with diet-induced or genetically induced obesity [19][45].
Recent studies confirm that macrophages release IL-1β under the stimuli of endocannabinoids, amyloid polypeptide and free fatty acids. In particular, the latter initially stimulates β cells to produce IL-1β and pro-inflammatory cytokines. Simultaneously, this phenomenon brings about an augmentation of nitric oxide levels as a result of the reduction in mitochondrial ATP. This event leads to β cell dysfunction and to a decreased production of insulin [19][20][45,46].
Hyperglycemia, in combination with insulin deficiency, leads to an impairment of both innate and adaptive immune responses. First, the production of cytokines, such as IL-1β, interleukin 2 (IL-2) and IL-6, produced by peripheral blood mononuclear cells is reduced under high glucose conditions, causing a decreased action against pathogens [13][39]. Secondly, hyperglycemia is responsible for the impairment in the recruitment of leukocytes and CD8+ T cells in mice since the adhesion molecules seem to be less expressed. Third, the properties neutrophils, such as their opsonization, phagocytosis, NET traps and ROS production, are decreased. Fourth, macrophages have a reduced phagocytosis due to the impairment of complement receptors and Fcγ receptors on isolated monocytes, changing their phenotype in M2 in mice, which causes a weak immune response against pathogens [13][39].