1. Primary Mitochondrial Diseases
In the case of primary mitochondrial diseases, those caused by mutations in nDNA or mtDNA genes that code for mitochondrial proteins, the activation of mtUPR leads to increased ATP production, the assembly of OXPHOS-related subunits, improved mitochondrial antioxidant capacity, and overall recovery of mitochondrial function
[1][131]. For example, Perry et al. demonstrated that mtUPR activation using tetracyclines, especially doxycycline, improved the survival and proliferation of MELAS cybrids and benefitted Rieske disease (mitochondrial complex III knockout) mouse fibroblasts and
ND1 mutant cybrids under low-glucose conditions. They also tested the effect of other mtUPR activators such as pentamidine, known to inhibit mitochondrial translation, and retapamulin, which inhibits prokaryotic translation, and found that cell survival increased in all cellular models of mitochondrial diseases under nutrient stress conditions. Furthermore, the cited authors tested the effect of tetracyclines in a mouse model of Leigh syndrome lacking the mitochondrial complex I protein NDUFS4, resulting in improvements in body weight loss, longevity, and the amelioration of neurological decline
[2][132]. Subsequently, Suárez-Rivero et al. confirmed the positive effect of tetracyclines and the consequent activation of mtUPR in fibroblasts derived from patients with mutations in the
GFM1 gene
[3][133]. Suárez-Rivero et al. also described that the activation of mtUPR via pterostilbene and mitochondrial cofactors improved mitochondrial function in different cellular models of primary mitochondrial diseases, enhancing the sirtuin mtUPR and transcriptional canonical mtUPR axes
[4][134].
Nevertheless, it is important to consider that the use of antibiotics and, especially, tetracyclines, for the treatment of mitochondrial diseases is controversial
[5][135].
On the other hand, it has been reported that mtUPR activation in urine-derived stem cells obtained from patients with the m.3243A>G mutation characteristic of MELAS syndrome induced mitochondrial dysfunction, while its inhibition through silencing ATF5 resulted in an improvement in cellular pathophysiology via increasing mitochondrial membrane potential and reducing ROS levels
[6][136]. This difference in outcomes could be due to varying levels of heteroplasmy that occur when mitochondrial diseases are caused by mtDNA mutations. For this reason, further studies are needed to confirm the effect of mtUPR activation on primary mitochondrial diseases.
2. Cardiac and Metabolic Disorders
In cardiac disorders, the mtUPR plays a protective role
[7][137]. Wang et al. observed that treatment with oligomycin or doxycycline in mice was beneficial with respect to cardiac ischemia–reperfusion injury, reducing infarct size. However, for this treatment to be effective, ATF5 and therefore mtUPR activation was required
[8][138]. In addition, Smyrnias et al. proposed that mtUPR induction through six different methods, namely, treatment with paraquat, treatment with isoproterenol (a β-adrenoreceptor agonist), treatment with G-TPP (a chaperone inhibitor), mutation of the ornithine transcarbamylase protein, treatment with Olaparib (a PARP inhibitor), or treatment with nicotinamide, improved mitochondrial respiration in stressed cardiomyocytes in mice and attenuated contractile failure and hypertrophy
[9][139]. Similarly, Xu et al. demonstrated that choline treatment in a rat model of heart disease activated the sirtuin mtUPR axis and inhibited both myocardial metabolic dysfunction and myocyte hypertrophy
[10][140]. Likewise, Wang et al. showed that mtUPR induction via oligomycin had a protective role in septic cardiomyopathy induced by lipopolysaccharide, reducing cardiac dysfunction and mitochondrial damage
[11][124]. Furthermore, Zhang et al. reported that the activation of mtUPR via treatment with tetrahydrocurcumin in mice subjected to transverse aortic constriction surgery reduced cardiac hypertrophy and surgery-induced oxidative stress
[12][141]. Nevertheless, as Smyrnias et al. described, the activation of the mtUPR is not only beneficial when induced by external agents, as cells themselves activate this compensatory pathway to reduce damage. Myocardial cells derived from patients with aortic stenosis exhibited high levels of mtUPR markers such as ATF5, Hsp60, or LonP1. Those with even higher levels showed reduced rates of cardiomyocyte death, decreased levels of abnormal fibrosis, and fewer markers of cardiac damage in plasma
[9][139].
On the other hand, metabolic diseases are related to alterations in OXPHOS gene expression and general mitochondrial dysfunction
[13][142]. The most widely studied metabolic disease is diabetes, characterized by high levels of glucose in the blood and insulin resistance
[14][143]. Elevated blood sugar levels over time inhibit the function of chaperones and proteases, leading to the accumulation of aberrant proteins and protein aggregates
[15][144]. For this reason, the activation of the mtUPR could be beneficial in the treatment of diabetes. Wardelmann et al. observed that treatment with intranasal insulin activated mtUPR in insulin-deficient mice, improving mitochondrial function, inhibiting autophagy, and preventing weight gain under high-fat diet conditions
[16][145]. In addition, Lee et al. demonstrated that LonP1 protease deficiency promoted liver gluconeogenesis and insulin resistance, while its overexpression mitigated liver insulin resistance induced by treatment with cholesterol and palmitate in liver SK-HEP-1 cells derived from humans
[17][146]. Moreover, Wu et al. suggested that high-fat or high-glucose diets increased ClpP protease expression in pancreatic Min6 β-cells, leading to a decrease in cell apoptosis and ROS production, as well as the stabilization of insulin signaling
[18][147]. Also, Kleinridders et al. reported that patients and mice with type 2 diabetes had lower Hsp60 expression levels in the brain, and this phenomenon was associated with insulin resistance
[19][148].
mtUPR modulation also provides protective effects in the metabolic liver disease setting. These pathologies are associated with alterations in OXPHOS and elevated ROS production. Gariani et al. described that treatment with nicotinamide adenine dinucleotide in mouse models of fatty liver disease reverted non-alcoholic fatty liver disease by activating SIRT1 and SIRT3, ultimately increasing hepatic β-oxidation and OXPHOS activity
[20][149]. In addition, the induction of the sirtuin mtUPR axes via nicotinamide precursor supplementation attenuated body weight gain and fat mass among mice fed a high-fat diet
[21][150].
3. Cancer
Finally, mtUPR modulation has also been proposed as a therapeutic target for the treatment of different types of cancer. However, in this case, the therapies are not based on the activation of this stress response but on its inhibition, as it has been shown that mtUPR markers such as ATF5, Hsp60, Hsp70, ClpP, or LonP1 are upregulated in a wide variety of cancers. There are many studies on this subject, which can be consulted in the following references:
[22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184].
In conclusion, a substantial body of literature supports the modulation of mtUPR as a therapeutic target in the treatment of various diseases, all of which have mitochondrial dysfunction as a primary underlying cause. RWesearchers will emphasize the utilization of this approach in the context of ageing and age-related diseases, particularly neurodegenerative disorders. These conditions have a significant societal impact, with a growing number of affected individuals and a notable lack of curative treatments.
4. Ageing and Neurodegenerative Diseases
Neurodegenerative diseases encompass a diverse group of disorders characterized by the progressive and selective loss of anatomically or physiologically connected neuronal systems
[56][185].
A major risk factor for neurodegenerative disorders is the process of ageing. It has been hypothesized that mitochondria play a central role in this context by producing ROS and accumulating mutations in mtDNA, both of which are believed to accelerate the physiological ageing process
[57][14]. It is well established that during ageing, mtDNA mutations accumulate, contributing to the decline in mitochondrial function
[58][186]. mtDNA is more vulnerable to damage due to the absence of histones and other chromatin proteins as well as its proximity to sites of ROS production
[59][187]. Furthermore, ROS imbalance has a significant impact on cognitive loss associated with ageing
[60][188]. The brain is particularly susceptible to oxidative damage due to its high oxygen consumption rate, high concentration of extremely peroxidizable unsaturated fatty acids, and relative lack of antioxidant enzymes compared to other organs
[61][189]. Additionally, ageing is accompanied by a reduction in mitophagy, which leads to the accumulation of damaged mitochondria, increased oxidative stress, and the induction of apoptosis
[62][190].
Given the pivotal role of mitochondrial dysfunction in ageing, it is not surprising that mitochondrial quality control mechanisms play a crucial role in this process. Increasing mitochondrial biogenesis by activating SIRT1 has been shown to delay ageing and extend lifespan in animal models
[23][63][152,191]. In line with this, numerous studies suggest that the induction of the mtUPR can extend lifespan. For instance, the lifespan of a complex IV mtETC mutant
C. elegans specimen exceeded that of control worms, and this extension was attributed to the activation of mtUPR, as evidenced by elevated markers of this stress response, such as Hsp6 and Hsp10
[64][95]. Moreover, mtUPR activation through mitochondrial translation inhibition, resveratrol, or PARP inhibitors promoted longevity among worms
[65][192]. Moreover, supplementation with NAD
+ precursors or the inhibition of NAD
+-consuming enzymes also prolonged lifespan in
C. elegans [66][193]. Importantly,
C. elegans is not the sole model organism in which the activation of the mtUPR is beneficial for ageing. In mice, damage to mitochondrial ribosomes and the subsequent mtUPR induction also delayed ageing
[67][194]. Furthermore, in
D. melanogaster, mtUPR stimulation via the overexpression of the TRAP1 chaperone promoted both longevity and healthspan
[68][195]. This beneficial effect was also observed in the fungal ageing model
P. anserina, where the overexpression of the mitochondrial protease LonP1, a marker of mtUPR, extended lifespan
[69][196].
In summary, there is sufficient evidence to suggest that the activation of the mtUPR can be helpful in delaying ageing and prolonging lifespan and that it may also play a beneficial role in the treatment of neurodegenerative diseases, as discussed below.
4.1. Alzheimer’s Disease
Alzheimer’s Disease (AD) is the most prevalent form of neurodegeneration, affecting more than 47.5 million people worldwide. A
lzheimer’s D
isease (AD) is characterized by two neuropathological hallmarks: (1) neurofibrillary tangles formed by neurofilaments and hyperphosphorylated tau protein and (2) extracellular senile plaques formed via the accumulation of amyloid β (Aβ) peptide. Together, these hallmarks result in an irreversible loss of neurons, especially in the cortex and hippocampus, leading to memory and cognition impairments. The majority of AD cases occur sporadically among individuals over 65 years of age
[70][197].
Numerous studies support the role of mitochondrial dysfunction in the pathogenesis of AD. For instance, the treatment of pig neurons with hydrogen peroxide increased intracellular Aβ levels
[71][198], and treatment with CCCP caused intracellular Aβ accumulation in cultured astrocytes
[72][199]. It has been suggested that oxidative damage precedes Aβ deposition in animal models
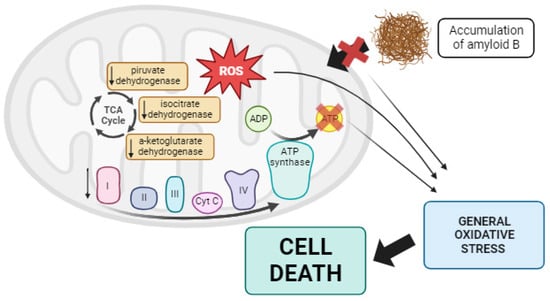
[73][200]. Additionally, post-mortem analysis of AD brains has revealed impaired activities of three essential mitochondrial enzymes: the pyruvate dehydrogenase complex, the α-ketoglutarate dehydrogenase complex, and isocitrate dehydrogenase
[74][75][76][201,202,203]. Moreover, defects in complexes of the mtETC have been reported in AD cases
[77][204]. These mtETC abnormalities reduced ATP production and increased ROS
[78][205]. Moreover, mitochondrial dysfunction, along with the resulting ROS overproduction, may alter amyloid precursor protein (APP) processing, leading to the intracellular accumulation of Aβ, a characteristic feature of AD
[71][198]. Aβ aggregation can block the import of nuclear-encoded mitochondrial proteins, causing oxidative damage to mitochondrial and cellular proteins, nucleic acids, and lipids. This sets off a vicious cycle, ultimately resulting in apoptosis and cell death
[79][80][206,207]. Moreover, mitochondrial size and shape are altered in AD patients, with mitochondria exhibiting an elongated, interconnected appearance referred to as “mitochondria on a string”
[81][208]. This is linked to disruptions in mitochondrial dynamics, particularly reduced mitochondrial fission and mitophagy, as noted by Zhang et al.
[82][209] (see
Figure 1).
Figure 1. Summary of mitochondrial dysfunction in AD. In Alzheimer’s disease, lower activity of some TCA enzymes such as pyruvate dehydrogenase, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase has been observed, along with alterations in the mtETC. These changes collectively result in reduced ATP production and the overproduction of ROS. Additionally, this promotes the aberrant processing of APP, leading to the accumulation of Aβ, which disrupts the import of mitochondrial proteins into the mitochondria. All of these factors contribute to generalized oxidative stress in the cell, ultimately causing cell death via apoptosis. Arrow: activation or induction of the subsequent process.
Considering the pivotal role of mitochondria in AD, the mtUPR could be a potential therapeutic target for this disease. Indeed, Beck et al. demonstrated an increase in the expression levels of mtUPR-associated proteins in the frontal cortex of sporadic AD patients
[83][210]. This finding has also been reported by other authors, such as Sorrentino et al., who observed an upregulation of mtUPR-associated genes in AD patients, mouse models, and
C. elegans as a protective response during disease progression
[84][211]. Moreover, in an AD
C. elegans model,
ATFS-1 depletion via RNAi led to severe cognitive impairment, reduced mitochondrial respiration, and exacerbated Aβ aggregation, highlighting the protective role of the mtUPR in this disease
[84][211]. Treatment with doxycycline or nicotinamide precursors to induce the mtUPR in the cited worm model and in mice improved overall health and lifespan by enhancing both animal motility and the clearance of Aβ peptides
[84][211]. These authors also tested the positive effect of doxycycline on SHSY5Y cells with an APP mutation, observing a reduction in Aβ deposits and an improvement in AD pathophysiology
[84][211]. Using the CL2006
C. elegans model of AD, which expresses the pathologic Aβ42, Regitz et al. demonstrated that treatment with resveratrol, a mtUPR activator, decreased Aβ aggregation and mitigated worm paralysis
[85][212]. Similarly, Pérez et al. observed the induction of the mtUPR in
pitrylisin metallopeptidase 1 (
PITRM1) mutant cortical neurons obtained from induced pluripotent stem cells, as shown by increased levels of ATF4, CHOP, Hsp60, ClpP, and LonP1. These mutant neurons exhibited increased levels of Aβ peptides without aggregation, possibly due to the protective role of the mtUPR. Inhibition of the mtUPR with ISRIB, an inhibitor of eif2α phosphorylation, resulted in the appearance of Aβ deposits and increased cell death
[86][213]. Likewise, the treatment of SHSY5Y cells with Aβ caused the activation of Hsp60, ClpP, and OMI. In fact, the activation of OMI protease was also detected in various human brain regions postmortem
[87][214]. In the brains of
APPsw/PS1De9 double transgenic mice, mtUPR induction was also demonstrated
[88][215]. Furthermore, the activation of the mtUPR via nicotinamide riboside or olaparib restored mitochondrial membrane potential and morphology, increased mtDNA content, reduced Aβ peptide aggregation, and improved muscle integrity and fitness in
C. elegans [89][216].
Considering all the studies published to date and the activation of the mtUPR in several Alzheimer’s disease models, it is evident that the induction of this mitochondrial stress response with external agents could be an effective strategy for the treatment of AD.
4.2. Parkinson’s Disease
Parkinson’s Disease (PD) is the most prevalent movement condition and the second-most common neurodegenerative disease, affecting more than 2% of the population over the age of sixty worldwide
[90][217]. This disorder is characterized by two hallmark features: (1) the presence of intraneuronal cytoplasmic aggregated α-synuclein protein inclusions known as Lewy bodies, and (2) a reduction in dopamine levels in the basal ganglia due to the death of dopaminergic neurons in the substantia nigra pars compacta, located in the midbrain
[91][218]. Several studies have suggested that mitochondrial dysfunction plays a role in the pathophysiology of PD. Mutations in genes such as
Parkin or
PINK1 are responsible for early-onset autosomal recessive PD, highlighting the importance of mitochondria in the disease’s etiology
[92][93][219,220]. Mutations in the
DJ-1 gene, which controls the NRF2 transcription factor involved in mitochondrial biogenesis
[94][221], and mutations in the OMI protease involved in the IMS-mtUPR
[95][222] have also been linked to PD.
However, most PD cases are not genetic but sporadic, and in the sporadic form of the disease, mitochondria are still implicated. Exposure to compounds such as MPTP or rotenone, which are inhibitors of mitochondrial complex I, can induce a parkinsonian phenotype, lead to the degeneration of dopaminergic neurons, and result in the occurrence of α-synuclein inclusions
[96][97][98][223,224,225]. Additionally, a reduction in complex I activity has been found in the brains of PD patients, establishing a link between complex I inhibition and sporadic PD
[99][226]. Moreover, inherited mtDNA mutations can also cause parkinsonism
[100][227], and PD patients have been found to have increased levels of somatic mtDNA deletions
[101][228]. Furthermore, according to some findings, oxidative stress and the presence of ROS may be among the primary causes of PD, as increased levels of oxidized lipids, proteins, and DNA have been detected in the substantia nigra of PD patients. In addition to ROS, reactive nitrogen species (RNS) also play an important role in PD. Nitric oxide (NO) is generated by nitric oxide synthase (NOS) and is present in high concentrations in cells and in the extracellular space surrounding dopaminergic neurons, as demonstrated in postmortem brain tissue from PD patients
[102][229]. NO can interfere with various enzymes and complexes I and IV, leading to increased ROS production. It can also cause lipid peroxidation and disrupt the proper functioning of proteins
[103][104][230,231].
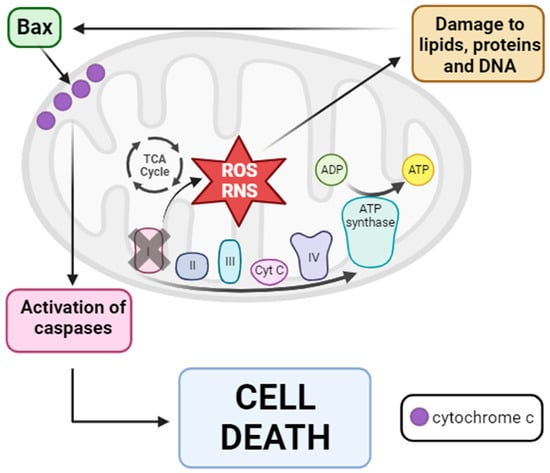
Based on these findings, a hypothesis regarding mitochondrial dysfunction in PD pathogenesis has been proposed. The inhibition of complex I disrupts electron flux through mtETC, leading to increased production of ROS and RNS. These reactive species damage different cellular components, including lipids, proteins, and DNAactivating proapoptotic pathways with the entry of Bax into the mitochondria. Bax subsequently causes the release of cytochrome c into the cytosol, ultimately activating the caspase signaling pathway and resulting in cell death via apoptosis
[105][232] (see
Figure 2).
Figure 2. Mitochondrial dysfunction hypothesis of PD pathogenesis. Inhibition of mitochondrial complex I provokes the disruption of mtETC with consequent ROS and RNS production. Then, these reactive species damage several cellular components such as lipids, proteins, and DNA, causing the entry of Bax from the cytosol into the OMM. Once in the OMM, Bax provokes the release of cytochrome c into the cytosol, leading to the activation of the caspase signaling pathway and finally cell death via apoptosis. Arrow: activation or induction of the subsequent process.
Given this hypothesis, it is not surprising that the activation of mitochondrial quality control mechanisms may act as a therapeutic target for Parkinson’s disease. Regarding the mtUPR, Cooper et al. showed that in mutant
PINK1,
Parkin, and
DJ-1 C. elegans PD models, mtUPR activation occurred, which protected worms from the death of dopaminergic neurons
[106][233]. Dastidar et al. reported that overexpression of 4E-BP1, an inhibitor of eukaryotic initiation factor 4E and protein translation, protected animals against neurotoxicity induced by rotenone and paraquat. This protection was attributed to the activation of the mtUPR, as indicated by increased levels of CHOP, ATF4, Hsp60, ClpP, and MnSOD
[107][234]. When CHOP was silenced, neuroprotection did not occur, and increased neuronal death was observed
[107][234]. Finally, Liu et al. proposed that, in a
D. melanogaster PD model, treatment with Ginseng, a Chinese herbal medicine, alleviated dopaminergic neuron death, improved locomotor function, and extended lifespan. These authors suggested that the beneficial effect of Ginseng was mediated via mtUPR enhancement, as they observed a significant induction of mtUPR-related genes such as Hsp60, Hsp70, and ClpP
[108][235].
Nonetheless, it is important to note that overactivation of the mtUPR, such as through the elimination of MTS of ATFS-1 in a
C. elegans PD model, provoked the death of dopaminergic neurons in a non-apoptotic manner, worsening the disease phenotype
[109][236]. For this reason, while mtUPR induction may be effective in the treatment of Parkinson’s disease, the levels of activation should be carefully controlled, as overactivation might have detrimental effects. Further research is needed to fully understand the optimal level of mtUPR activation for therapeutic benefit.
4.3. Huntington’s Disease
In Huntington’s Disease (HD), the role of mitochondrial dysfunction has also been proposed. HD is characterized by the progressive loss of GABAergic medium spiny neurons in the striatum, although degeneration is also observed in other brain regions such as the thalamus, the subthalamic nucleus, white matter, or the cerebellum
[110][237]. This disorder affects 1 in 10,000 people worldwide
[111][238]. Unlike Alzheimer’s and Parkinson’s diseases, where most cases are sporadic, the cause of HD is genetic
[112][239]. This disease is caused by a CAG triplet expansion in the
huntingtin gene, resulting in an expansion of polyglutamine in the N-terminal end of the protein. A number greater than 40 repeats is considered pathogenic, while the normal number is less than 36
[113][240].
It has been reported that mutant huntingtin (mtHtt) causes defects in mitochondria, leading to neurotoxicity, neuronal dysfunction, and finally cell death
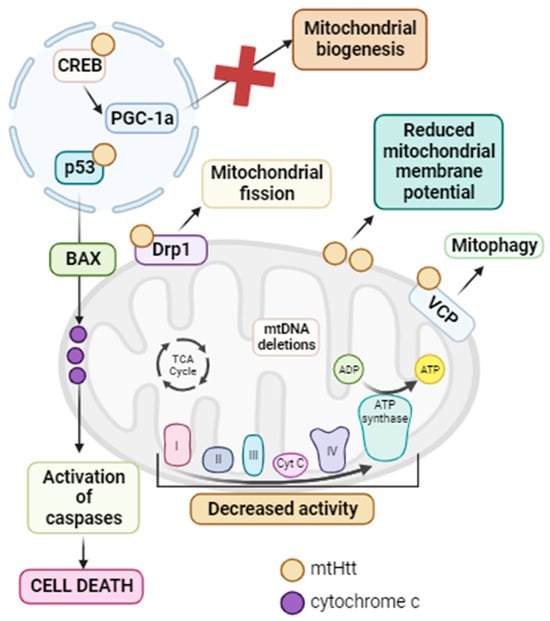
[114][241]. Indeed, it has been demonstrated that mtHtt interacts with several proteins. In the nucleus, mtHtt binds to p53, a tumor suppressor that, in response to stress, activates Bax and thus cell death via apoptosis
[115][242]. The inhibition of p53 in
D. melanogaster and
M. musculus HD models prevented cellular dysfunction and neurodegeneration
[116][243]. In addition, mtHtt interacts with CREB and CREB binding protein (CBP), inhibiting their function and therefore mitochondrial biogenesis
[117][244]. In mitochondria, mtHtt activates the Drp1 protein, leading to mitochondrial fragmentation and general mitochondrial dysfunction
[118][245]. Another protein with which mtHtt interacts is valosin-containing protein (VCP), an ATPase associated with OMM protein turnover and parkin-dependent mitophagy. As a result, mtHtt–VCP interaction promotes increased mitophagy and neuronal degeneration associated with mitochondrial dysfunction
[119][246]. The involvement of mitochondrial dysfunction has been confirmed, as decreased mitochondrial complex I, II, III, and IV activity has been reported in the brains of HD patients
[114][120][241,247], along with impaired mitochondrial respiration and ATP production
[121][248]. Moreover, the inhibition of mitochondrial complex II by 3-nitropropionic acid or malonate leads to a pathological phenotype that resembles HD
[122][249]. Impaired succinate dehydrogenase activity has also been observed in postmortem HD brains, along with reduced mitochondrial membrane potential in HD patients’ muscles and lymphoblasts
[114][241]. This could be due to the interaction of mutant huntingtin with the OMM, which causes mitochondrial membrane potential reduction and potentiates mitochondrial permeability transition
[123][250]. Finally, mtDNA deletions have been identified in HD patients during disease progression
[124][251].
In summary, mtHtt interacts with a large number of proteins and mitochondrial structures such as the OMM. This interaction results in a reduction in mitochondrial biogenesis and membrane potential. It also leads to the opening of the mitochondrial permeability transition pore, calcium homeostasis failure, the induction of mitophagy, and mitochondrial fission. Additionally, there is a decrease in mitochondrial complex activity, elevated ROS production, and, ultimately, cell death, provoking the neurodegeneration characteristic of HD
[125][252]. For this reason, the activation of mitochondrial quality control mechanisms and especially the mtUPR could be a potential treatment since mtHtt undergoes misfolding and proteolytic cleavage, resulting in N-terminal fragments that can form aggregates (see
Figure 3).
Figure 3. Summary of mitochondrial dysfunction in HD. mtHtt interacts with a large number of proteins. In the nucleus, it interacts with p53, promoting the activation of Bax with the subsequent release of cytochrome c from the mitochondria and the activation of caspases, ultimately leading to cell death via apoptosis. Additionally, in this same cellular compartment, mtHtt interacts with CREB, inhibiting it and thus suppressing PGC-1α and mitochondrial biogenesis. In the mitochondria, mtHtt interacts with Drp1, causing its activation and consequent mitochondrial fragmentation. Moreover, mtHtt binds to the OMM, leading to a decrease in mitochondrial membrane potential. It also interacts with VCP, activating mitophagy. All of these processes, along with the decreased activity of mtETC complexes and the accumulation of mtDNA deletions, contribute to the mitochondrial dysfunction characteristic of HD. Arrow: activation or induction of the subsequent process.
Accordingly, Fu et al. related that ABCB10, a component of the mtUPR pathway, had decreased transcript and protein expression levels in an HD mouse model and in dermal fibroblasts of two HD patients
[126][253]. Overexpression of ABCB10 in striatal cells of an HD mouse model induced CHOP expression and reduced fragmented mitochondria, ROS production, and cell death via apoptosis
[126][253]. The protein and mRNA levels of two markers of the mtUPR, Hsp60 and ClpP, were significantly reduced in fibroblasts derived from two HD patients, suggesting that mtHtt inhibited mtUPR activation in sufferers of HD
[126][253]. Moreover, Hernández et al. observed that the ATF5 transcription factor, the master regulator of the mtUPR, was sequestered in mtHtt aggregates in HD mice,
C. elegans, and patients, mainly in the cortex and striatum in the case of mice and patients
[127][254]. Almeida et al. also observed decreased levels of mtUPR markers in PC12 cells expressing mtHtt
[128][255]. On the other hand, Naia et al. demonstrated that SIRT3 expression levels and activity were elevated in several in vitro and in vivo models of HD
[129][256]. However, this upregulation is not sufficient to counteract the toxic effects of mtHtt. Overexpression of SIRT3 in HD mouse and
D. melanogaster models, as well as in HD-patient-derived fibroblasts, resulted in increased mitochondrial membrane potential, the elevation of mitochondrial complex activity, and decreased mitochondrial fragmentation, leading to the improvement of HD pathophysiology
[129][256]. Furthermore, these authors also tested the effect of resveratrol in an HD mouse model, with the result being the activation of mitochondrial biogenesis through SIRT1 induction
[130][257]. In the striatal cells derived from this HD mouse model, the use of nicotinamide induced the activation of sirtuin mtUPR, provoking an increase in mitochondrial antioxidant capacity with a consequent increase in mitochondrial membrane potential
[130][257]. Likewise, treatment with both resveratrol and nicotinamide in HD-derived lymphoblasts restored mitochondrial respiration, with increased mitochondrial biogenesis and ATP production
[130][257]. The positive effect of resveratrol and nicotinamide was also tested in the HD mouse model, where the authors observed that the restoration of mitochondrial function alleviated neurodegeneration signs and improved motor function
[130][257]. Furthermore, Fu et al. showed that the activation of mitochondrial SIRT3 via treatment with ε-viniferin in a PC12 HD cell model reduced oxidative stress, promoted mitochondrial biogenesis, and improved mitochondrial dysfunction
[131][258].
In conclusion, mitochondrial dysfunction is strongly associated with the pathogenesis of HD. mtHtt interacts with many proteins, among which is ABCB10, involved in the mtUPR, or CREB implicated in mitochondrial biogenesis, resulting in an impairment in mitochondrial quality control mechanisms. Therefore, the use of compounds that induce these pathways may be a potential therapeutic option for the treatment of HD.
4.4. Amyotrophic Lateral Sclerosis
Amyotrophic Lateral Sclerosis (ALS) is a fatal neurodegenerative disorder characterized by the progressive loss of upper and lower motor neurons, mainly in the spinal cord, brainstem, and motor cortex. With an incidence of approximately 2.5/per 100,000 people worldwide, ALS is the third-most-common neurodegenerative disease after AD and PD
[132][259]. While the majority of ALS cases are sporadic, 10% have a genetic cause. Since the discovery of the first gene associated with ALS,
superoxide dismutase 1 (
SOD1)
[133][260], more than 20 other genes have been reported to correlate with its pathogenesis, including
matrin 3 (
MATR3),
coiled-coil-helix-coiled-coil-helix domain-containing 10 (
CHCHD10),
fused in sarcoma/translocated in liposarcoma (
FUS),
chromosome 9 open reading frame 72 (
C9orf72),
TAR DNA binding protein 43 (
TDP-43), and
OPTN, among others
[134][261].
Several studies have proposed a strong association between mitochondrial function and ALS pathogenesis. In the case of familial ALS, mutations in the
SOD1 gene alter its antioxidant capacity, leading to ROS accumulation that inactivates axonal transport, induces axonal degeneration, and causes SOD1 to bind to Bcl-2, inhibiting Bcl-2’s anti-apoptotic activity. This promotes cytochrome C release and mitochondrially initiated caspase activation. In addition, altered calcium homeostasis in mitochondria due to
SOD1 mutations provokes the activation of apoptotic pathways and motor neuron death
[135][262].
C9orf72 mutations are also associated with mitochondrial dysfunction since C9orf72 is an IMM-associated protein. C9orf72 regulates complex I assembly by interacting with the IMM-domain-containing 1 (TIMMDC1) protein. When C9orf72 is mutated, the result is reduced mitochondrial complex I activity, provoking mitochondrial dysfunction and ALS motor symptoms
[136][263]. On the other hand, mutations in
TDP-43 cause an alteration in mitochondrial dynamics, with an increased expression of FIS1 and Drp1 and decreased levels of Mfn1, leading to mitochondrial fragmentation, which affects mitochondrial function, including mtETC activity, ROS production, and calcium homeostasis, resulting in neuronal apoptosis
[137][264]. In addition, mutations in
OPTN cause a disruption in mitophagy since this protein is the primary receptor for PINK1/Parkin-mediated mitophagy. This causes an accumulation of damaged mitochondria and, once again, mitochondrial dysfunction, with a consequent cascade of signaling that eventually leads to neuronal death
[138][265].
MATR3 mutations have been found in four families with ALS. This protein has RNA- and DNA-binding domains that regulate gene expression, and it forms a complex with TDP43 and FUS, contributing to the mitochondrial dysfunction described above
[139][266]. Similarly,
CHCHD10 mutations cause mitochondrial dynamics and cellular bioenergetics disturbances, as ALS patients with mutations in this gene present a fragmented mitochondrial network
[140][267]. Moreover, the CHCHD10 protein interacts with TDP43 in the nucleus, but when it is mutated, the interaction does not occur, and TDP43 translocates to the cytosol, causing synaptic damage
[141][268]. Finally, in the case of
FUS mutations, the protein encoded by this gene forms aggregates in the cytosol of motor neurons. Additionally, it has been described that mutant FUS protein targets mRNAs encoding mitochondrial respiratory chain proteins in the nucleus and in the mtDNA, causing OXPHOS deficiency, ROS overproduction, and general mitochondrial dysfunction
[142][269].
It is important to emphasize that in all these cases, there is an aggregation of the mutated protein inside the mitochondrion, which disrupts OXPHOS and leads to ROS accumulation, Ca
2+ dyshomeostasis, mitochondrial dynamics alterations, mitophagy failure, and mitochondrial dysfunction. These alterations alter axonal transport, induce axonal degeneration, and ultimately lead to neuronal apoptosis
[143][270].
Regarding sporadic cases, even though they do not follow a family history, mutations in some of the ALS-related genes have been found in most cases. In Table 1, researchers summarized the principal genes associated with ALS and their pathogenic mechanisms.
Table 1.
Summary of the main genes related to ALS and their pathogenic mechanisms. It is important to note that mitochondrial dysfunction is present in all cases.
It is therefore important to consider the role of mitochondria, and most notably mitochondrial dysfunction and oxidative stress, in the pathogenic mechanisms of ALS.
Interestingly, the mtUPR is also associated with ALS. Wang et al. observed mtUPR activation in TDP-43 ALS cellular and animal models. Moreover, they described that TDP-43 interacted with LonP1, leading to its degradation. LonP1 overexpression protected cells from TDP-43-induced neurotoxicity, reducing mitochondrial damage and neurodegeneration. In contrast, inhibiting this protease decreased cell viability, suggesting a protective role of the mtUPR in TDP-43 pathogenesis
[144][271].
In addition, Deng et al. demonstrated that mutant FUS accumulated within mitochondria, where FUS aggregates interacted with the ATP5B subunit of the ATP synthase complex. This interaction reduced mitochondrial ATP synthesis in HEK293 cell lines expressing mutant FUS and in a FUS-ALS
D. melanogaster model. As a result, mitochondrial impairment occurred, and the mtUPR was activated, evidenced by the upregulation of mtUPR-associated genes such as ATF5, LonP1, Hsp60, and Hsp70 in both ALS cellular and animal models
[145][272]. However, in contrast to the TDP-43 mutation case, LonP1 overexpression in the FUS-ALS
D. melanogaster model exacerbated retinal degeneration, with increased neurodegeneration, while silencing mtUPR-related genes ameliorated mutant FUS-induced neurodegeneration
[145][272].
On the other hand, Zhou et al. reported that mtUPR activation in an ALS mouse model with an
SOD1 gene mutation, achieved via treatment with nicotinamide precursors, decreased mitochondrial dysfunction and SOD1 aggregates and improved adult neurogenesis
[146][273]. Furthermore, Straub et al. observed elevated mtUPR marker levels for both the transcriptional canonical and sirtuin mtUPR axes in fibroblasts derived from mutant
CHCHD10 patients when cultured under galactose stress conditions
[147][274]. Finally, Riar et al. described that in a mouse model of ALS carrying the G93A-mutation in the SOD1 protein, IMS-mtUPR axis activation was induced when mutant SOD1 accumulated in the IMS. Interestingly, the genetic ablation of ERα induced the activation of the transcriptional canonical mtUPR axis, suggesting that in the absence of ERα, cells induced the CHOP/ATF/ATF5 axis as a compensatory mechanism
[148][275]. It is worth noting that the activation of both axes of the mtUPR was found to be sex-specific, with significantly higher levels of induction observed in female mice compared to male mice (for which the levels were low)
[148][275].
In conclusion, the role of the mtUPR as a therapeutic target in ALS remains complex. Its activation shows varied effects, with some cases indicating positive outcomes due to the aggregation of mutated proteins, while others demonstrate deleterious effects, as observed with LonP1 overexpression. A potential avenue worth exploring is the induction of the sirtuin axis, as suggested by the positive effects seen in the treatment with nicotinamide precursors. However, it is important to note that this observation is based on limited studies, and further research is essential to elucidate the full therapeutic potential of mtUPR pathways in ALS.
4.5. Friedreich’s Ataxia (FA)
3.4.5. Friedreich’s Ataxia (FA)
The implications of mitochondrial dysfunction and the mtUPR have also been described in Friedreich’s Ataxia (FA), a rare disease arising from a GAA repeat expansion in the
frataxin (
FXN) gene, which encodes the mitochondrial FXN protein involved in iron-sulfur cluster biosynthesis
[149][276]. Consequently, decreased protein expression levels of FXN result in mitochondrial dysfunction, characterized by reduced OXPHOS, ROS overproduction, abnormal mitochondrial morphology, and Ca
2+ homeostasis alterations
[150][277]. It has been reported that transcriptional canonical mtUPR axis markers such as ATF4 and CHOP were upregulated in a mouse model of FA
[151][278].