Muscular dystrophies are a heterogeneous group of genetic muscle-wasting disorders that are subdivided based on the region of the body impacted by muscle weakness as well as the functional activity of the underlying genetic mutations. A common feature of the pathophysiology of muscular dystrophies is chronic inflammation associated with the replacement of muscle mass with fibrotic scarring. With the progression of these disorders, many patients suffer cardiomyopathies with fibrosis of the cardiac tissue. Anti-inflammatory glucocorticoids represent the standard of care for Duchenne muscular dystrophy, the most common muscular dystrophy worldwide; however, long-term exposure to glucocorticoids results in highly adverse side effects, limiting their use. Thus, it is important to develop new pharmacotherapeutic approaches to limit inflammation and fibrosis to reduce muscle damage and promote repair.
- muscular dystrophy
- dystrophin
- dysferlin
- Emery-Dreifuss
- dystroglycan
- LAMA2
- FSHD
1. Introduction
2. Duchenne and Becker Muscular Dystrophy
Duchenne muscular dystrophy (DMD), the most common and severe form of MD, is caused by mutations in the dystrophin (DMD) gene (Figure 1). This gene is located on the X chromosome [2], and DMD affects 1 in 3600 males born [3]. Clinically, DMD is characterized by muscle weakness and wasting during early childhood, with symptoms appearing by age 3. Initially, these patients have difficulty or pain with movement, frequent falls, a waddling gait, and delayed growth [4]. There is a loss of ambulation in childhood to early teens and death in early adulthood [5][6]. DMD patients also suffer significant cardiac complications; approximately 90% will progress to dilated cardiomyopathy marked by inflammatory cell infiltration, fibrosis, and myocardial cell death, leading to early mortality [7][8]. In contrast, Becker MD (BMD), the second most common form of muscular dystrophy, also caused by mutations in DMD, is found in 1.5–3.6/100,000 males globally. BMD is a milder disease, and patients typically are identified at 11–25 years of age; however, they may have much later ages of onset. BMD patients develop progressive weakness in the muscles of the hips, thighs, pelvis, and shoulders and generally have a nearly normal lifespan unless they experience cardiac failure [9].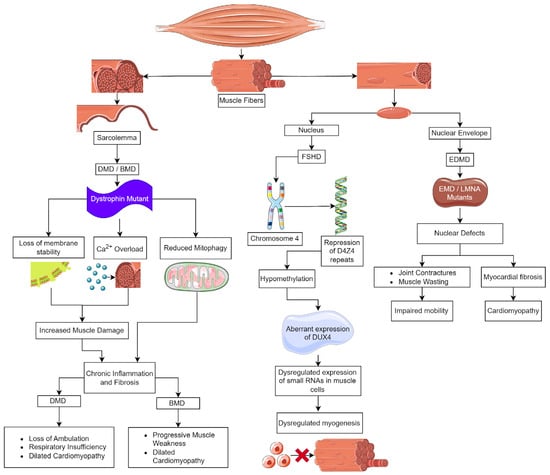
2.1. Inhibition of Inflammation and Fibrosis
Edasalonexent (CAT-1004, is a small molecule NF-κB inhibitor that combines structural elements of salicylic acid and docosahexenoic acid [20]. A phase 3 placebo-controlled clinical trial of this drug in DMD boys (aged 4–8 years) found it to be safe and tolerated, but functional data from the North Star Ambulatory Assessment (NSAA) criteria showed a nonsignificant trend of improvement in younger patients (<6 years) (NCT03703882) [1]. Edasalonexent may prolong ambulation and delay respiratory and cardiac problems when prescribed for very young patients [20]. Unfortunately, Catabasis Pharmaceuticals stopped development of the drug. Vamorolone, or VBP15, is a 21-aminosteroid or lazaroid, which is a glucocorticoid steroid that has a delta 9,11 modification of the backbone [21]. Vamorolone stabilizes membranes by reducing lipid peroxidation and inhibiting NF-kB-mediated inflammation. Importantly, vamorolone limits adverse steroid side effects because it binds the glucocorticoid receptor without activating gene expression via the glucocorticoid response element (GRE) [22][23][24][25]. Pamrevlumab is a monoclonal antibody specific for CTGF, which regulates pro-fibrotic pathways and is upregulated in DMD patients along with TGF-β [16]. FibroGen assessed pamrevlumab (FG-3019) in two clinical trials in DMD patients, examining its effect on limb muscles and respiratory function. Their phase 3 trial on upper limb performance in non-ambulatory DMD patients found that while pamrevlumab was safe and tolerated, it did not meet its primary endpoint (NCT04371666), whereas the phase 2 trial on respiratory function is ongoing (NCT02606136). In response to muscle injury, the urokinase-type plasminogen activator and its receptor (uPA/uPAR) form a complex that leads to ECM breakdown, the release of TGF-β, and subsequent macrophage invasion, all necessary processes in the course of normal muscle repair. Normally, active TGF-β is limited by the feedback of plasminogen activator inhibitor 1 (PAI-1) on uPA[26] [33] [34]. In the context of dystrophic muscle, this system is dysregulated, resulting in highly elevated levels of TGF-β that contribute to fibrosis and macrophage infiltration [35],[36]. It was proposed that the chronic inflammation of DMD is linked to persistent uPA/uPAR activity and disruption of the negative feedback loop between TGF-β and PAI-1 [37]. Serp1 is a protein derived from Myxoma virus that mimics the activity of PAI-1, inhibiting uPA and reducing TGF-β. In preclinical studies, a pegylated Serp1, PEGSerp1 was administered to Mdx/Utrn-/- (DKO) mice, whose phenotype recapitulates the time course and pathology of human DMD [37]. In DKO diaphragms, PEGSerp1 treatment resulted in increased fiber diameters, decreased fibrotic area, and decreased pro-inflammatory macrophage invasion [37]. This demonstrated that inhibiting the uPA/uPAR/TGF-β pathway will potentially reduce fibrosis and inflammation. Serp-1 represents a new biological approach for advances in therapeutics to address inflammation and fibrosis. [27]Since TGF-β has a multifactorial role in signaling during regeneration, therapeutic strategies focused on limiting fibrosis often target this protein. One such strategy is a monoclonal antibody that targets latent transforming growth factor beta binding protein 4 (LTBP4), a matrix-embedded protein that sequesters TGF-β in the ECM and modulates its availability. LTBP4 also binds other members of the TGF-β superfamily, including myostatin, which may aid its efficacy [2826]. The antibody is directed against the hinge region of LTBP4; when bound, it maintains the protein in a closed conformation, securing TGF-β and reducing the amount that is freely acting in muscle. Treatment of mdx mice for 6 months resulted in increased fiber size and less fibrosis, and there was a synergistic effect when combined with corticosteroids [2826]. This preclinical study is encouraging for DMD patients, most of whom are treated with corticosteroids.2.2. Calcium Homeostasis[29]
2.2. Calcium Homeostasis
Disruption of Ca2+ homeostasis in muscular dystrophy has been identified as a key contributor to skeletal myofiber and myocardial cell death [3027][3128][3229]. Elevated cytosolic Ca2+ activates calcium-sensitive proteases, calpain and phospholipase A2, in myofibers, resulting in proteolysis and membrane damage. Though initially attributed to Ca2+ influx through microtears of the sarcolemma, it is clear that increased Ca2+ in the cytosol is caused by activation of channels in the membrane of the sarcoplasmic reticulum (SR), such as the ryanodine receptors (RyR1 and RyR2) and the sarcolemma, such as the transient receptor potential canonical channels (TRPC), voltage-gated Na+ channels (Nav1.4 and Nav1.5), and ionotropic purinoreceptors (P2 × 7). Further, in DMD, the reduced activity of the SERCA1 and SERCA2 channels that mediate Ca2+ uptake in the SR and the mitochondrial Ca2+ uniporter (MCU) also contributes to the loss of calcium homeostasis [3330][3431]. It has been hypothesized that the DGC serves as a scaffold for the assembly of the channels and facilitates the activation of mechanosensitive channels. Ion channels that promote Ca2+ influx through the sarcolemma cluster with the DGC by direct interactions with α-syntrophins [3532]. In the absence of dystrophin, the integrity of many channels is disrupted, including TRPCs, store-operated Ca2+ entry (SOCE) channels, and voltage-gated Na+ channels, leading to an excessive influx of Ca2+ from the environment. Dystrophic muscle in mice, rats, and DMD patients overexpress TRPC1, TRPC3, and TRPC6, further increasing the Ca2+ influx into the muscle [3633][3734]. The activity of TRPC3 and TRPC6 is increased by phosphorylation by Ca2+ calmodulin-dependent kinase (CaMKII) and ERK1/2 and reduced N-linked glycosylation [3835][3936][4037]. The importance of TRPC6 in DMD fibrosis and muscle weakening was demonstrated by treating Mdx/Utrn−/− mice, a severe model of DMD, orally with the TRPC6 antagonist BI 749327 [4138]. Reduced TRPC6 activity in the double knock-out mice resulted in an extended lifespan, increased muscle and heart function, and reduced fibrosis in the gastrocnemius, diaphragm, and ventricle, as compared to untreated controls. Similar anti-fibrotic outcomes have been observed in mice for cardiac and renal disease [4239].2.3. Inhibition of Oxidative Stress
3. Emery–Dreifuss Muscular Dystrophy (EDMD)
Emery–Dreifuss muscular dystrophy (EDMD) is a neuromuscular disorder characterized by a combination of skeletal muscle weakness, joint contractures, and cardiac abnormalities [5653]. EDMD is rare; it has a prevalence estimated at 3 to 4 cases per million. Falling under the spectrum of laminopathies, EDMD is caused by mutations in at least 10 genes encoding nuclear envelope proteins. The most common types are caused by mutations in emerin (EMD) and lamin A (LMNA) [5653][5754][5855]. EMD has an X-linked recessive inheritance [5855], it encodes emerin, a nuclear membrane protein found at the interface of the nucleus with the cytoplasm. LMNA is inherited in an autosomal dominant fashion, and it codes for lamin A and C, which are type V intermediate filaments. The lamins polymerize under the inner nuclear membrane to form the nuclear lamina [5956][6057]. EDMD patients have diverse clinical presentations, making it challenging to diagnose and manage [5653][55][58][61]. Patients with EDMD have three main sets of symptoms, which consist of early contractures, progressive muscle weakness and atrophy, and cardiac abnormalities. Joint contractures are a hallmark symptom of EDMD, causing multi-joint stiffness, reduced mobility, and functional impairment, most often involving the neck, elbows, and heel. Spinal rigidity may also affect posture, flexibility, and swallowing [5653][5855]. Progressive muscle weakness and wasting primarily affect the scapulo-humero-peroneal muscles in children, which impairs mobility and strength [5653]. In the nucleus, emerin binds to lamin A, actin, and nuclear myosin, and as a complex, it regulates chromatin dynamics, acts as a mechanosensor for the nucleus, and participates in the regulation of gene expression [6259][6360][6461][6562]. EDMD pathophysiology involves disruptions in the nuclear envelope, resulting in loss of nuclear membrane integrity, heterochromatin decondensation, nucleoplasm leakage, chromatin detachment from the nuclear lamina, pseudoinclusions, and nuclear fragmentation (Figure 1) [5653]. These defects contribute to cellular dysfunction, especially in tissues like cardiac and skeletal muscles that undergo mechanical stress [5855]. Histologically, the heart demonstrates myocardial fibrosis and fibrolipomatosis, and skeletal muscle has dystrophic changes, including variation in fiber size, increased number of myofibers with centrally localized nuclei, increased endomysial connective tissue, and the presence of necrotic fibers [5855].EDMD Treatment
EDMD is a challenging and genetically complex disorder, necessitating ongoing research to advance our understanding of the disease and develop effective therapeutic strategies [5855][6663]. Timely cardiac interventions, regular monitoring, and supportive care are also essential in improving the prognosis and quality of life for individuals with EDMD [5653]. Currently, the therapeutic approaches for EDMD patients are limited to the alleviation of symptoms, slowing disease progression, and improving the overall quality of life [5653]. Much like DMD and BMD patients, glucocorticoids are prescribed to limit inflammation [5754]. Due to side effects associated with long-term use of glucocorticoids, anti-inflammatory and anti-fibrotic agents are being examined in mouse models for EDMD.4. Facioscapulohumeral Muscular Dystrophy (FSHD)
Facioscapulohumeral muscular dystrophy (FSHD) is the third most prevalent type of MD, following myotonic dystrophy type 1 and DMD, estimated to affect 1 in 15,000 to 1 in 20,000 people [6764]. FSHD is inherited in an autosomal dominant manner; however, approximately 30% are de novo cases. Additionally, there is a high frequency of somatic mosaicism [6865]. FSHD typically presents with weakness in one or more of these facial muscles, the stabilizers of the scapula, or the dorsiflexors of the foot. Muscle weakness is slowly progressive and, with time, can involve the axial and respiratory muscles and those of the lower limbs [6966]. Histopathological studies of human FSHD biopsies have shown that there are regenerating myofibers of varying sizes; inflammation and fibrosis that is both endomysial and perivascular were prominent, and there was significant fat replacement of the muscle fibers [7067]. A two-year prospective study of FSHD patients found that inflammation was mild in the early stages, but its presence was related to an increased rate of muscle degradation and fat infiltration [7168]. When examining RNAseq and microarray data from FSHD biopsies, it was noted that inflammatory genes have increased levels of expression [7269]. FSHD myoblasts have increased sensitivity to oxidative stress and are prone to apoptosis because double homeobox protein 4 (DUX4) affects mitochondrial function. DUX4 is the gene affected in FSHD patients [7370][7471]. DUX4 is expressed in pre-implantation embryos, and Dux binding sites are found in the control regions of genes involved in early genome activation [7269][7370]. DUX4 is important in early embryos for embryonic genome activation (EGA). DUX4 triggers this process and subsequently becomes inactive [7572][7673][7774][7875][7976]. Postnatally, DUX4 is expressed at low levels in the testis and the thymus. Ectopic expression in skeletal muscle postnatally results in FSHD [8077][8178][8279]. The human DUX4 gene is located on chromosome 4 in a region known as D4Z4, which consists of 11 to more than 100 repeated 3300 DNA base pair segments. Each of the repeated segments in the D4Z4 region contains a copy of the DUX4 gene; the copy closest to the end of chromosome 4 is called DUX4, while the other copies are referred to as “DUX4-like” or DUX4L [8380][8481]5. Limb-Girdle Muscular Dystrophy
Limb-girdle muscular dystrophy (LGMD) is the largest group of muscular dystrophies. They were originally defined by the progressive wasting of skeletal muscles of the pelvic and pectoral girdles. These dystrophies show significant variation in the onset of the disease, degree of wasting and inclusion of cardiomyopathy, cardiac arrhythmias, and respiratory failure [8582]. There are 24 genetic subtypes based on disease phenotype and mutations: (i) sarcoglycan complex (LGMD2C-F); (ii) glycosylation/α-dystroglycan complex (LGMD2I, LGMD2K, LGMD2M, LGMD2N, LGMD2O, LGMD2P, LGMD2S, LGMD2T, LGMD2U, LGMD2Z); (iii) sarcomeric proteins (LGMD1A, LGMD1D, LGMD1E, LGMD2A, LGMD2G, LGMD2J, LGMD2Q, and LGMD2R); (iv) signal transduction (LGMD1C, LGMD2P, LGMD2W); and (v) membrane trafficking and repair (LGMD1C, LGMD1F, LGMD2B, LGMD2L). These disorders are relatively rare compared to DMD, with individual estimated prevalences of 0.01–0.60 cases per 100,000 persons [8683]. The rarity of these disorders limits the ability to carry out clinical trials to examine the effectiveness of therapeutic strategies discovered in mice [8784].
5.1. Sarcoglycanopathy
Sarcoglycan consists of four subunits, α, β, γ, and δ, that form an integral membrane protein complex that is a subunit of the DGC in skeletal muscle. This protein complex is essential for the structural stability of the sarcolemma in both skeletal and cardiac muscle (Figure 2). Genetic mutations of any sarcoglycan subunit are inherited in an autosomal dominant fashion and result in sarcoglycanopathy, which has a similar phenotype to DMD. In sarcoglycanopathy, or LGMD2, the average age at onset of muscle weakness is 6–8 years, with loss of ambulation at 12–16 years old. Similar to DMD, cardiomyopathy and respiratory complications following the loss of ambulation are commonly observed [82][85][8886][89]. At the tissue level, muscle weakness is associated with chronic inflammation and the replacement of muscle fibers with fat depositions and fibrosis [9087].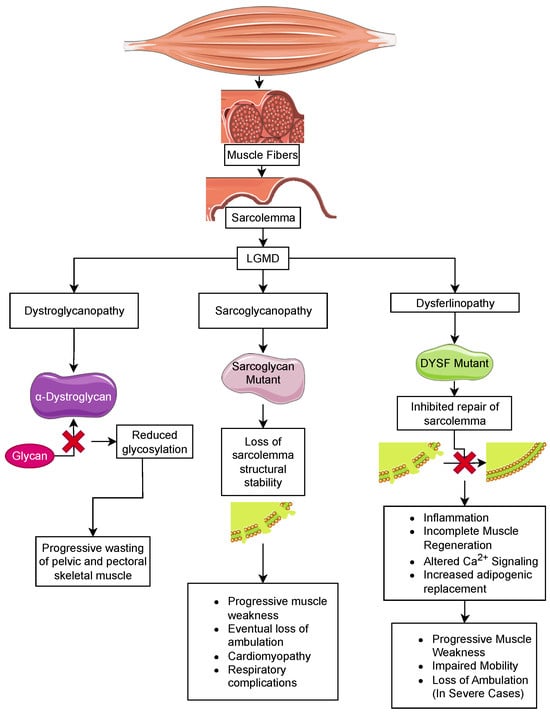
5.2. Dystroglycanopathies
6. Congenital Muscular Dystrophy
6.1. LAMA2-Related Muscular Dystrophy
MDC1A patients display hypotonia and muscle wasting at birth and with progressive spinal deformities and contractures of the major limb joints in early childhood [10299][103100]. These patients can suffer from neuropathies under the age of 1 and can experience cerebral atrophy and seizures in older individuals [104101][105102][106103]. In severe cases, the loss of muscle strength complicates swallowing and breathing, and death due to respiratory insufficiency can occur in the first decade, though their usual life expectancy is the thirties [107104][108105]. Inflammation and fibrosis play prominent roles in the early onset of muscle weakness in MDC1A [109106]. Individuals experience a burst of inflammation in their skeletal muscle as early as a couple of months after birth. This leads to the expansion of the interstitial ECM between myofibers, resulting in fibrosis. Unlike DMD, where fibrosis builds over time with repeated rounds of muscle breakdown and repair, fibrosis in MDC1A appears early and is maintained [109106]. Fibrosis in MDC1A muscle has been linked to the TGF-β and renin-angiotensin signaling pathways. TGF-β is a primary regulator of fibrotic deposition via its roles in promoting the expression of ECM proteins and the conversion of satellite cells to a fibroblastic lineage [16][110107]. Therapeutic strategies designed to reduce fibrosis in MDC1A patients have largely focused on reducing TGF-β levels. One approach that has shown promise is the AT1R blocker losartan. Though initially approved by the FDA to control hypertension, it effectively reduced fibrosis in the dyW/dyW and dy2J/dy2J by disruption of Ang II signaling through AT1R, which inhibits the conversion of TGF-β to its active form [111108][112109][113110][114111]. Losartan treatment also decreased ERK phosphorylation and fibrosis in dyW/dyW mice [111108]. The FDA-approved serine/threonine kinase inhibitor vemurafenib effectively ameliorates fibrosis in dy3K/dy3K mice [115112]. Treatment resulted in a reduction in TGF-β and mTORC1/p70S6K signaling. Much like losartan, vemurafenib was able to block the pro-fibrotic pathway but was unable to promote muscle repair. This raises concerns about its use as a solo therapy. In addition to blocking the TGF-β signaling pathway that directly induces fibrosis and inflammation, other research groups have explored the therapeutic benefits of blocking secondary events associated with muscle pathology in MDC1A patients and mouse models. Perhaps the most promise has been reported with omigapil, an inhibitor of the pro-apoptotic protein, Bax, and the GAPDH-Siah apoptosis cascade [116113][117114] The pathology in Lama2-deficient mice appears to be exacerbated by the upregulation of lysosome-associated degradation pathways in the muscle [118115][119116]. Consistent with this, inhibition of the ubiquitin-proteasome system by treatment of dy3K/dy3K mice with MG-132 or bortezomib increased muscle strength and mobility and reduced muscle fibrosis while extending the mouse’s lifespan [118115][120117]. In wildtype mice, Lama2 acts as an inhibitor of autophagy through the inhibition of the class III PI3K Vps34.6.2. Collagen VI-Related Dystrophies
ColVI is a beaded fibril consisting of three chains encoded by COL6A1, COL6A2, and COL6A3. The ColVI collagen fibers form a mesh-like network associated with the ECM of basement membranes, where it can modify the nature of the structure based on binding to glycoproteins, proteoglycans, and glycosaminoglycans [121118]. They serve important roles in protecting cells from compressive forces while facilitating the interaction between cells and ECM proteins. Further, ColVI participates in the regulation of autophagy through direct interaction with integral membrane proteins on adjacent cells (β1 integrin, TEM8, and VEGFR2) [122119][123120][124121]. Mutations in COL6A1, COL6A2, and COL6A3 lead to a spectrum of congenital muscular dystrophies that range from UCMD at the severe end of the spectrum to Bethlem myopathy at the mild end [121118]. UCMD patients experience congenital hypotonia and contractures in joints of the extremities. The histopathology associated with muscle weakness in ColVI-related dystrophies includes muscle fibers of various sizes and a significant increase in interstitial fibrosis [125122]. As with MDC1A, fibrosis accumulation is immediate and does not build with time, as seen with DMD. Using Col61A-deficient mice as a model, fibrosis was found to be related to overactivation of mesenchymal stem cells that have been converted to the fibroblast lineage [125122].7. Conclusions
Advances in genetic, genomic, and proteomic technologies have resulted in an acceleration in the unraveling of the molecular mechanisms underlying the histopathology of more than 50 types of muscular dystrophy and opened the door to an exploration of alternative therapeutic approaches to glucocorticoid suppression of fibrosis, fat accumulation, and chronic inflammation associated with most muscular dystrophies. Strategies with broad promise are those that reduce proinflammatory NF-κB and TNF-α signaling. Studies in mouse models have shown success in ameliorating the pathology upstream of their expression through antioxidants (e.g., COX enzyme inhibitor) [126123], suppression of the inflammasome (e.g., P2X7 antagonists) [127124] or directly through the inhibition of NF-κB activity (e.g., vamorolone) [128125]. Alternatively, inhibition of the Akt/mTOR signaling pathway by rapamycin suppresses the immune response to injury by reducing the proliferation of immune cells [129126]. This has shown potential for ameliorating fibrosis in Lmna- and Ftkn-deficient mice [10198][130127][131128][132129]. Recent reports predict that direct suppression of fibrotic deposition in fibroblasts at the site of injury by disrupting uPA/uPAR signaling via Serp-1 represents a strategy downstream of chronic inflammation to reduce fibrosis [133130][134131][135132].References
- Benarroch, L.; Bonne, G.; Rivier, F.; Hamroun, D. The 2023 version of the gene table of neuromuscular disorders. Neuromuscul. Disord. 2023, 33, 76–117.
- Roberts, R.; Cole, C.; Hart, K.; Bobrow, M.; Bentley, D. Rapid carrier and prenatal diagnosis of Duchenne and Becker muscular dystrophy. Nucleic Acids Res. 1989, 17, 811.
- Mah, J.K.; Korngut, L.; Dykeman, J.; Day, L.; Pringsheim, T.; Jette, N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 2014, 24, 482–491.
- Yiu, E.M.; Kornberg, A.J. Duchenne muscular dystrophy. J. Paediatr. Child Health 2015, 51, 759–764.
- Jennekens, F.G.; ten Kate, L.P.; de Visser, M.; Wintzen, A.R. Diagnostic criteria for Duchenne and Becker muscular dystrophy and myotonic dystrophy. Neuromuscul. Disord. 1991, 1, 389–391.
- Shirikova, N.; Niggli, E. Cardiac phenotype of Duchenne Muscular Dystrophy: Insights from cellular studies. J. Mol. Cell. Cardiol. 2013, 58, 217–224.
- Klingler, W.; Jurkat-Rott, K.; Lehmann-Horn, F.; Schleip, R. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. 2012, 31, 184–195.
- Mavrogeni, S.; Papavasiliou, A.; Spargias, K.; Constandoulakis, P.; Papadopoulos, G.; Karanasios, E.; Georgakopoulos, D.; Kolovou, G.; Demerouti, E.; Polymeros, S.; et al. Myocardial inflammation in Duchenne Muscular Dystrophy as a precipitating factor for heart failure: A prospective study. BMC Neurol. 2010, 10, 33–39.
- Aartsma-Rus, A.; Van Deutekom, J.C.; Fokkema, I.F.; Van Ommen, G.J.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144.
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517.
- Doorenweerd, N.; Mahfouz, A.; van Putten, M.; Kaliyaperumal, R.; t’Hoen, P.A.C.; Hendriksen, J.G.M.; Aartsma-Rus, A.M.; Verschuuren, J.G.M.; Niks, E.H.; Reinders, M.J.T.; et al. Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy. Sci. Rep. 2017, 7, 12575.
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002, 82, 291–329.
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239.
- Duan, D.; Goemans, N.; Takeda, S.; Mervuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13.
- Gloss, D.; Moxley, R.T.; Ashwal, S.; Oskoui, M. Practice guideline update summary: Corticosteroid treatment of Duchenne muscular dystrophy. Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2016, 86, 465–472.
- Song, Y.; Yao, S.; Liu, Y.; Long, L.; Yang, H.; Li, Q.; Liang, J.; Li, X.; Lu, Y.; Zhu, H.; et al. Expression levels of TGF-β1 and CTGF are associated with the severity of Duchenne muscular dystrophy. Exp. Ther. Med. 2017, 13, 1209–1214.
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93.
- Kourakis, S.; Timpani, C.A.; Campelj, D.G.; Hafner, P.; Gueven, N.; Fischer, D.; Rybalka, E. Standard of care versus new-wave corticosteroids in the treatment of Duchenne muscular dystrophy: Can we do better? Orphanet J. Rare Dis. 2021, 16, 117.
- Mah, J.K.; Clemens, P.R.; Guglieri, M.; Smith, E.C.; Finkel, R.S.; Tulinius, M.; Nevo, Y.; Ryan, M.M.; Webster, R.; Castro, D.; et al. Efficacy and Safety of Vamorolone in Duchenne Muscular Dystrophy: A 30-Month Nonrandomized Controlled Open-Label Extension Trial. JAMA Netw. Open 2022, 5, e2144178.
- Finkel, R.S.; McDonald, C.M.; Lee Sweeney, H.; Finanger, E.; Neil Knierbein, E.; Wagner, K.R.; Mathews, K.D.; Marks, W.; Statland, J.; Nance, J.; et al. A Randomized, Double-Blind, Placebo-Controlled, Global Phase 3 Study of Edasalonexent in Pediatric Patients with Duchenne Muscular Dystrophy: Results of the PolarisDMD Trial. J. Neuromuscul. Dis. 2021, 8, 769–784.
- Reeves, E.K.; Hoffman, E.P.; Nagaraju, K.; Damsker, J.M.; McCall, J.M. VBP15: Preclinical characterization of a novel anti-inflammatory delta 9, 11 steroid. Bioorg. Med. Chem. 2013, 21, 2241–2249.
- Bracken, M.B.; Shepard, M.J.; Holford, T.R.; Leo-Summers, L.; Aldritch, E.F.; Fazl, M.; Fehlings, M.; Herr, D.L.; Hitchon, P.W.; Marshall, L.F. Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury. Results of the National Acute Spinal Cord Injury Randomized Controlled Trial. National Acute Spinal Cord Injury Study. JAMA 1997, 277, 1597–1604.
- Cahill, L.; Hall, E.D. Is it time to resurrect “lazaroids”? J. Neurosci. Res. 2017, 95, 17–20.
- Heier, C.R.; Damsker, J.M.; Yu, Q.; Dillingham, B.C.; Huynh, T.; Van der Meulen, J.H.; Sali, A.; Miller, B.K.; Phadke, A.; Sheffer, L.; et al. VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol. Med. 2013, 5, 1569–1585.
- Baudy, A.R.; Reeves, E.K.; Damsker, J.M.; Heier, C.; Garvin, L.M.; Dillingham, B.C.; McCall, J.; Rayavarapu, S.; Wang, Z.; Vandermeulen, J.H. Δ-9,11 modification of glucocorticoids dissociates nuclear factor-κB inhibitory efficacy from glucocorticoid response element-associated side effects. J. Pharmacol. Exp. Ther. 2012, 343, 225–232.
- Suelves, M.; López-Alemany, R.; 2002Lluı́s, F.; Aniorte, G.; Serrano, E.; Parra, M.; Carmeliet, P.; Muñoz-Cánoves, P. Plasmin Activity Is Required for Myogenesis in Vitro and Skeletal Muscle Regeneration in Vivo.. Blood. 2002, 99, 2835–2844 .Demonbreun, A.R.; Fallon, K.S.; Oosterbaan, C.C.; Vaught, L.A.; Reiser, N.L.; Bogdanovic, E.; Velez, M.P.; Salamone, I.M.; Page, P.G.T.; Hadhazy, M.; et al. Anti-latent TGF-β binding protein 4 antibody improves muscle function and reduces muscle fibrosis in muscular dystrophy. Sci. Trans. Med. 2021, 13, eabf0376.
- Suelves, M.; López-Alemany, R.; Lluı́s, F.; Aniorte, G.; Serrano, E.; Parra, M.; Carmeliet, P.; Muñoz-Cánoves, P. Plasmin Activity Is Required for Myogenesis in Vitro and Skeletal Muscle Regeneration in Vivo.. Blood . 2002, 99, 2835–2844 .Vallejo-Illarramendi, A.; Toral-Ojeda, I.; Aldanondo, G.; López de Munain, A. Dysregulation of calcium homeostasis in muscular dystrophies. Expert Rev. Mol. Med. 2014, 16, e16.
- Demonbreun, A.R.; Fallon, K.S.; Oosterbaan, C.C.; Vaught, L.A.; Reiser, N.L.; Bogdanovic, E.; Velez, M.P.; Salamone, I.M.; Page, P.G.T.; Hadhazy, M.; et al. Anti-latent TGF-β binding protein 4 antibody improves muscle function and reduces muscle fibrosis in muscular dystrophy. Sci. Trans. Med. 2021, 13, eabf0376. Burr, A.R.; Molkentin, J.D. Genetic evidence in the mouse solidifies the calcium hypothesis of myofiber death in muscular dystrophy. Cell Death Differ. 2015, 22, 1402–1412.
- 32. Suelves, M.; López-Alemany, R.; Lluı́s, F.; Aniorte, G.; Serrano, E.; Parra, M.; Carmeliet, P.; Muñoz-Cánoves, P. Plasmin Activity Is Required for Myogenesis in Vitro and Skeletal Muscle Regeneration in Vivo. Blood 2002, 99, 2835–2844 doi:10.1152/ajpcell.00568.2007. 33. Suelves, M. The Plasminogen Activation System in Skeletal Muscle Regeneration: Antagonistic Roles of Urokinase-Type Plasminogen Activator (Upa) and Its Inhibitor (PAI-1). Front. Biosci. 2005, 10, 2978, doi: 10.2741/1754 34. Suelves, M.; Vidal, B.; Serrano, A.L.; Tjwa, M.; Roma, J.; López-Alemany, R.; Luttun, A.; de Lagrán, M.M.; Díaz, M.À.; Jardí, M. et al. UPA Deficiency Exacerbates Muscular Dystrophy in MDX Mice. J. Cell. Biol. 2007, 178, 1039–1051, doi:10.1083/jcb.2007051235. Lluís, F.; Roma, J.; Suelves, M.; Parra, M.; Aniorte, G.; Gallardo, E.; Illa, I.; Rodríguez, L.; Hughes, S.M.; Carmeliet, P. et al. Urokinase-Dependent Plasminogen Activation Is Required for Efficient Skeletal Muscle Regeneration in Vivo. Blood 2001, 97, 1703–1711, doi:10.1182/blood.v97.6.1703 36. Ardite, E.; Perdiguero, E.; Vidal, B.; Gutarra, S.; Serrano, A.L.; Muñoz-Cánoves, P. PAI-1–Regulated MiR-21 Defines a Novel Age-Associated Fibrogenic Pathway in Muscular Dystrophy. J. Cell Biol. 2012, 196, 163–175, doi:10.1083/jcb.201105013 37. Andre, A.B.; Zhang, L.; Nix, J.D.; Elmadbouly, N.; Lucas, A.R.; Wilson-Rawls, J.; Rawls, A. Myxomavirus Serp-1 Protein Ameliorates Inflammation in a Mouse Model of Duchenne Muscular Dystrophy. Biomedicines, 2022, 10, 1154. doi: 10.3390/biomedicines10051154Mareedu, S.; Million, E.D.; Duan, D.; Babu, G.J. Abnormal calcium handling in Duchenne Muscular Dystrophy: Mechanisms and potential therapies. Front. Physiol. 2021, 12, 647010.
- Vallejo-Illarramendi, A.; Toral-Ojeda, I.; Aldanondo, G.; López de Munain, A. Dysregulation of calcium homeostasis in muscular dystrophies. Expert Rev. Mol. Med. 2014, 16, e16. Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364.
- Burr, A.R.; Molkentin, J.D. Genetic evidence in the mouse solidifies the calcium hypothesis of myofiber death in muscular dystrophy. Cell Death Differ. 2015, 22, 1402–1412. Schneider, J.S.; Shanmugam, M.; Gonzalez, J.P.; Lopez, H.; Gordan, R.; Fraidenraich, D.; Babu, G.J. Increased sarcolipin expression and decreased sarco(endo)plasmic reticulum Ca2+ uptake in skeletal muscles of mouse models of Duchenne muscular dystrophy. J. Muscle Res. Cell Motil. 2013, 34, 349–356.
- Mareedu, S.; Million, E.D.; Duan, D.; Babu, G.J. Abnormal calcium handling in Duchenne Muscular Dystrophy: Mechanisms and potential therapies. Front. Physiol. 2021, 12, 647010. Constantin, B. Dystrophin complex functions as a scaffold for signalling proteins. Biochim. Biophys. Acta 2014, 1838, 635–642.
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. Hammers, D.W.; Sleeper, M.M.; Forbes, S.C.; Shima, A.; Walter, G.A.; Sweeney, H.L. Tadalafil treatment delays the onset of cardiomyopathy in dystrophin-deficient hearts. J. Am. Heart Assoc. 2016, 5, e003911.
- Schneider, J.S.; Shanmugam, M.; Gonzalez, J.P.; Lopez, H.; Gordan, R.; Fraidenraich, D.; Babu, G.J. Increased sarcolipin expression and decreased sarco(endo)plasmic reticulum Ca2+ uptake in skeletal muscles of mouse models of Duchenne muscular dystrophy. J. Muscle Res. Cell Motil. 2013, 34, 349–356. Creisméas, A.; Gazaille, C.; Bourdon, A.; Lallemand, M.A.; François, V.; Allais, M.; Ledevin, M.; Larcher, T.; Toumaniantz, G.; Lafoux, A.; et al. TRPC3, but not TRPC1, as a good therapeutic target for standalone or complementary treatment of DMD. J. Transl. Med. 2021, 19, 519.
- Constantin, B. Dystrophin complex functions as a scaffold for signalling proteins. Biochim. Biophys. Acta 2014, 1838, 635–642. Shi, J.; Geshi, N.; Takahashi, S.; Kiyonaka, S.; Ichikawa, J.; Hu, Y.; Mori, Y.; Ito, Y.; Inoue, R. Molecular determinants for cardiovascular TRPC6 channel regulation by Ca2+/calmodulin-dependent kinase II. J. Physiol. 2013, 591, 2851–2866.
- Hammers, D.W.; Sleeper, M.M.; Forbes, S.C.; Shima, A.; Walter, G.A.; Sweeney, H.L. Tadalafil treatment delays the onset of cardiomyopathy in dystrophin-deficient hearts. J. Am. Heart Assoc. 2016, 5, e003911. Dietrich, A.; y Schnitzler, M.M.; Emmel, J.; Kalwa, H.; Hofmann, T.; Gudermann, T. N-linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J. Biol. Chem. 2003, 278, 47842–47852.
- Creisméas, A.; Gazaille, C.; Bourdon, A.; Lallemand, M.A.; François, V.; Allais, M.; Ledevin, M.; Larcher, T.; Toumaniantz, G.; Lafoux, A.; et al. TRPC3, but not TRPC1, as a good therapeutic target for standalone or complementary treatment of DMD. J. Transl. Med. 2021, 19, 519. Shen, B.; Kwan, H.Y.; Ma, X.; Wong, C.O.; Du, J.; Huang, Y.; Yao, X. cAMP activates TRPC6 channels via the phosphatidylinositol 3-kinase (PI3K)-protein kinase B (PKB)-mitogen-activated protein kinase kinase (MEK)-ERK1/2 signaling pathway. J. Biol. Chem. 2011, 286, 19439–19445.
- Shi, J.; Geshi, N.; Takahashi, S.; Kiyonaka, S.; Ichikawa, J.; Hu, Y.; Mori, Y.; Ito, Y.; Inoue, R. Molecular determinants for cardiovascular TRPC6 channel regulation by Ca2+/calmodulin-dependent kinase II. J. Physiol. 2013, 591, 2851–2866. Lin, B.L.; Shin, J.Y.; Jeffreys, W.P.; Wang, N.; Lukban, C.A.; Moorer, M.C.; Velarde, E.; Hanselman, O.A.; Kwon, S.; Kannan, S.; et al. Pharmacological TRPC6 inhibition improves survival and muscle function in mice with Duchenne muscular dystrophy. JCI Insight 2022, 7, e158906.
- Dietrich, A.; y Schnitzler, M.M.; Emmel, J.; Kalwa, H.; Hofmann, T.; Gudermann, T. N-linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J. Biol. Chem. 2003, 278, 47842–47852. Lin, B.L.; Matera, D.; Doerner, J.F.; Zheng, N.; Del Camino, D.; Mishra, S.; Bian, H.; Zeveleva, S.; Zhen, X.; Blair, N.T.; et al. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc. Natl. Acad. Sci. USA 2019, 116, 10156–10161.
- Shen, B.; Kwan, H.Y.; Ma, X.; Wong, C.O.; Du, J.; Huang, Y.; Yao, X. cAMP activates TRPC6 channels via the phosphatidylinositol 3-kinase (PI3K)-protein kinase B (PKB)-mitogen-activated protein kinase kinase (MEK)-ERK1/2 signaling pathway. J. Biol. Chem. 2011, 286, 19439–19445. Bellinger, A.M.; Reiken, S.; Carlson, C.; Mongillo, M.; Liu, X.; Rothman, L.; Matecki, S.; Lacampagne, A.; Marks, A.R. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 2009, 15, 325–330.
- Lin, B.L.; Shin, J.Y.; Jeffreys, W.P.; Wang, N.; Lukban, C.A.; Moorer, M.C.; Velarde, E.; Hanselman, O.A.; Kwon, S.; Kannan, S.; et al. Pharmacological TRPC6 inhibition improves survival and muscle function in mice with Duchenne muscular dystrophy. JCI Insight 2022, 7, e158906. Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442.
- Lin, B.L.; Matera, D.; Doerner, J.F.; Zheng, N.; Del Camino, D.; Mishra, S.; Bian, H.; Zeveleva, S.; Zhen, X.; Blair, N.T.; et al. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc. Natl. Acad. Sci. USA 2019, 116, 10156–10161. Voit, A.; Patel, V.; Pachon, R.; Shah, V.; Bakhutma, M.; Kohlbrenner, E.; McArdle, J.J.; Dell’Italia, L.J.; Mendell, J.R.; Xie, L.H.; et al. Reducing sarcolipin expression mitigates Duchenne muscular dystrophy and associated cardiomyopathy in mice. Nat. Commun. 2017, 8, 1068.
- Bellinger, A.M.; Reiken, S.; Carlson, C.; Mongillo, M.; Liu, X.; Rothman, L.; Matecki, S.; Lacampagne, A.; Marks, A.R. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 2009, 15, 325–330. Wasala, N.B.; Yue, Y.; Lostal, W.; Wasala, L.P.; Niranjan, N.; Hajjar, R.J.; Babu, G.J.; Duan, D. Single SERCA2a therapy ameliorated dilated cardiomyopathy for 18 months in a mouse model of duchenne muscular dystrophy. Mol. Ther. 2020, 28, 845–854.
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. Reid, A.L.; Alexander, M.S. The Interplay of Mitophagy and Inflammation in Duchenne Muscular Dystrophy. Life 2021, 11, 648.
- Voit, A.; Patel, V.; Pachon, R.; Shah, V.; Bakhutma, M.; Kohlbrenner, E.; McArdle, J.J.; Dell’Italia, L.J.; Mendell, J.R.; Xie, L.H.; et al. Reducing sarcolipin expression mitigates Duchenne muscular dystrophy and associated cardiomyopathy in mice. Nat. Commun. 2017, 8, 1068. Bround, M.J.; Havens, J.R.; York, A.J.; Sargent, M.A.; Karch, J.; Molkentin, J.D. ANT-dependent MPTP underlies necrotic myofiber death in muscular dystrophy. Sci. Adv. 2023, 9, eadi2767.
- Wasala, N.B.; Yue, Y.; Lostal, W.; Wasala, L.P.; Niranjan, N.; Hajjar, R.J.; Babu, G.J.; Duan, D. Single SERCA2a therapy ameliorated dilated cardiomyopathy for 18 months in a mouse model of duchenne muscular dystrophy. Mol. Ther. 2020, 28, 845–854. Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831.
- Reid, A.L.; Alexander, M.S. The Interplay of Mitophagy and Inflammation in Duchenne Muscular Dystrophy. Life 2021, 11, 648. Reutenauer, J.; Dorchies, O.M.; Patthey-Vuadens, O.; Vuagniaux, G.; Ruegg, U.T. Investigation of Debio 025, a cyclophilin inhibitor, in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. Br. J. Pharmacol. 2008, 155, 574–584.
- Bround, M.J.; Havens, J.R.; York, A.J.; Sargent, M.A.; Karch, J.; Molkentin, J.D. ANT-dependent MPTP underlies necrotic myofiber death in muscular dystrophy. Sci. Adv. 2023, 9, eadi2767. Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447.
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831. Wissing, E.R.; Millay, D.P.; Vuagniaux, G.; Molkentin, J.D. Debio-025 is more effective than prednisone in reducing muscular pathology in mdx mice. Neuromuscul. Disord. 2010, 20, 753–760.
- Reutenauer, J.; Dorchies, O.M.; Patthey-Vuadens, O.; Vuagniaux, G.; Ruegg, U.T. Investigation of Debio 025, a cyclophilin inhibitor, in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. Br. J. Pharmacol. 2008, 155, 574–584. Lawler, J.M. Exacerbation of pathology by oxidative stress in respiratory and locomotor muscles with Duchenne muscular dystrophy: Oxidative stress and DMD pathology. J. Physiol. 2011, 589, 2161–2170.
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. Buyse, G.M.; Goemans, N.; van den Hauwe, M.; Thijs, D.; de Groot, I.J.M.; Schara, U.; Ceulemans, B.; Meier, T.; Mertens, L. Idebenone as a novel, therapeutic approach for Duchenne muscular dystrophy: Results from a 12 month, double-blind, randomized placebo-controlled trial. Neuromuscul. Disord. 2011, 21, 396–405.
- Wissing, E.R.; Millay, D.P.; Vuagniaux, G.; Molkentin, J.D. Debio-025 is more effective than prednisone in reducing muscular pathology in mdx mice. Neuromuscul. Disord. 2010, 20, 753–760. Rovira Gonzalez, Y.I.; Moyer, A.L.; LeTexier, N.J.; Bratti, A.D.; Feng, S.; Peña, V.; Sun, C.; Pulcastro, H.; Liu, T.; Iyer, S.R. Mss51 deletion increases endurance and ameliorates histopathology in the mdx mouse model of Duchenne muscular dystrophy. FASEB J. 2021, 35, e21276.
- Lawler, J.M. Exacerbation of pathology by oxidative stress in respiratory and locomotor muscles with Duchenne muscular dystrophy: Oxidative stress and DMD pathology. J. Physiol. 2011, 589, 2161–2170. Madej-Pilarczyk, A. Clinical aspects of Emery-Dreifuss muscular dystrophy. Nucleus 2018, 9, 268–274.
- Buyse, G.M.; Goemans, N.; van den Hauwe, M.; Thijs, D.; de Groot, I.J.M.; Schara, U.; Ceulemans, B.; Meier, T.; Mertens, L. Idebenone as a novel, therapeutic approach for Duchenne muscular dystrophy: Results from a 12 month, double-blind, randomized placebo-controlled trial. Neuromuscul. Disord. 2011, 21, 396–405. Muchir, A.; Worman, H.J. Emery–Dreifuss muscular dystrophy: Focal point nuclear envelope. Curr. Opin. Neurol. 2019, 32, 728–734.
- Rovira Gonzalez, Y.I.; Moyer, A.L.; LeTexier, N.J.; Bratti, A.D.; Feng, S.; Peña, V.; Sun, C.; Pulcastro, H.; Liu, T.; Iyer, S.R. Mss51 deletion increases endurance and ameliorates histopathology in the mdx mouse model of Duchenne muscular dystrophy. FASEB J. 2021, 35, e21276. Heller, S.A.; Shih, R.; Kalra, R.; Kang, P.B. Emery-Dreifuss muscular dystrophy. Muscle Nerve 2020, 61, 436–448.
- Madej-Pilarczyk, A. Clinical aspects of Emery-Dreifuss muscular dystrophy. Nucleus 2018, 9, 268–274. Solovei, I.; Wang, A.S.; Thanisch, K.; Schmidt, C.S.; Krebs, S.; Zwerger, M.; Cohen, T.V.; Devys, D.; Foisner, R.; Peichl, L.; et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 2013, 152, 584–598.
- Muchir, A.; Worman, H.J. Emery–Dreifuss muscular dystrophy: Focal point nuclear envelope. Curr. Opin. Neurol. 2019, 32, 728–734. Bertrand, A.T.; Brull, A.; Azibani, F.; Benarroch, L.; Chikhaoui, K.; Stewart, C.L.; Medalia, O.; Ben Yaou, R.; Bonne, G. Lamin A/C Assembly Defects in LMNA-Congenital Muscular Dystrophy Is Responsible for the Increased Severity of the Disease Compared with Emery–Dreifuss Muscular Dystrophy. Cells 2020, 9, 844.
- Heller, S.A.; Shih, R.; Kalra, R.; Kang, P.B. Emery-Dreifuss muscular dystrophy. Muscle Nerve 2020, 61, 436–448. Meinke, P.; Kerr, A.R.W.; Czapiewski, R.; de Las Heras, J.I.; Dixon, C.R.; Harris, E.; Kölbel, H.; Muntoni, F.; Schara, U.; Straub, V.; et al. A multistage sequencing strategy pinpoints novel candidate alleles for Emery-Dreifuss muscular dystrophy and supports gene misregulation as its pathomechanism. eBioMedicine 2020, 51, 102587.
- Solovei, I.; Wang, A.S.; Thanisch, K.; Schmidt, C.S.; Krebs, S.; Zwerger, M.; Cohen, T.V.; Devys, D.; Foisner, R.; Peichl, L.; et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 2013, 152, 584–598. Guilluy, C.; Osborne, L.D.; Van Landeghem, L.; Sharek, L.; Superfine, R.; Garcia-Mata, R.; Burridge, K. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell Biol. 2014, 16, 376–381.
- Bertrand, A.T.; Brull, A.; Azibani, F.; Benarroch, L.; Chikhaoui, K.; Stewart, C.L.; Medalia, O.; Ben Yaou, R.; Bonne, G. Lamin A/C Assembly Defects in LMNA-Congenital Muscular Dystrophy Is Responsible for the Increased Severity of the Disease Compared with Emery–Dreifuss Muscular Dystrophy. Cells 2020, 9, 844. Le, H.Q.; Ghatak, S.; Yeung, C.Y.; Tellkamp, F.; Günschmann, C.; Dieterich, C.; Yeroslaviz, A.; Habermann, B.; Pombo, A.; Niessen, C.M.; et al. Mechanical regulation of transcription controls Polycomb-mediated gene silencing during lineage commitment. Nat. Cell Biol. 2016, 18, 864–875.
- Meinke, P.; Kerr, A.R.W.; Czapiewski, R.; de Las Heras, J.I.; Dixon, C.R.; Harris, E.; Kölbel, H.; Muntoni, F.; Schara, U.; Straub, V.; et al. A multistage sequencing strategy pinpoints novel candidate alleles for Emery-Dreifuss muscular dystrophy and supports gene misregulation as its pathomechanism. eBioMedicine 2020, 51, 102587. Holaska, J.M.; Wilson, K.L. An emerin “proteome”: Purification of distinct emerin-containing complexes from HeLa cells suggests molecular basis for diverse roles including gene regulation, mRNA splicing, signaling, mechanosensing, and nuclear architecture. Biochemistry 2007, 46, 8897–8908.
- Guilluy, C.; Osborne, L.D.; Van Landeghem, L.; Sharek, L.; Superfine, R.; Garcia-Mata, R.; Burridge, K. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell Biol. 2014, 16, 376–381. Pradhan, R.; Ranade, D.; Sengupta, K. Emerin modulates spatial organization of chromosome territories in cells on softer matrices. Nucleic Acids Res. 2018, 46, 5586.
- Le, H.Q.; Ghatak, S.; Yeung, C.Y.; Tellkamp, F.; Günschmann, C.; Dieterich, C.; Yeroslaviz, A.; Habermann, B.; Pombo, A.; Niessen, C.M.; et al. Mechanical regulation of transcription controls Polycomb-mediated gene silencing during lineage commitment. Nat. Cell Biol. 2016, 18, 864–875. Wang, S.; Peng, D. Cardiac Involvement in Emery-Dreifuss Muscular Dystrophy and Related Management Strategies. Int. Heart J. 2019, 60, 12–18.
- Holaska, J.M.; Wilson, K.L. An emerin “proteome”: Purification of distinct emerin-containing complexes from HeLa cells suggests molecular basis for diverse roles including gene regulation, mRNA splicing, signaling, mechanosensing, and nuclear architecture. Biochemistry 2007, 46, 8897–8908. Hamel, J.; Johnson, N.; Tawil, R.; Martens, W.B.; Dilek, N.; McDermott, M.P.; Heatwole, C. Patient-reported symptoms in facioscapulohumeral muscular dystrophy (PRISM-FSHD). Neurology 2019, 93, e1180–e1192.
- Pradhan, R.; Ranade, D.; Sengupta, K. Emerin modulates spatial organization of chromosome territories in cells on softer matrices. Nucleic Acids Res. 2018, 46, 5586. Chen, T.H.; Wu, Y.Z.; Tseng, Y.H. Early-onset infantile facioscapulohumeral muscular dystrophy: A timely review. Int. J. Mol. Sci. 2020, 21, 7783.
- Wang, S.; Peng, D. Cardiac Involvement in Emery-Dreifuss Muscular Dystrophy and Related Management Strategies. Int. Heart J. 2019, 60, 12–18. Steel, D.; Main, M.; Manzur, A.; Muntoni, F.; Munot, P. Clinical features of fascioscapulohumeral muscular dystrophy 1 in childhood. Dev. Med. Child Neurol. 2019, 61, 964–971.
- Hamel, J.; Johnson, N.; Tawil, R.; Martens, W.B.; Dilek, N.; McDermott, M.P.; Heatwole, C. Patient-reported symptoms in facioscapulohumeral muscular dystrophy (PRISM-FSHD). Neurology 2019, 93, e1180–e1192. Wang, L.H.; Tawil, R. Facioscapulohumeral Dystrophy. Curr. Neurol. Neurosci. Rep. 2016, 16, 66.
- Chen, T.H.; Wu, Y.Z.; Tseng, Y.H. Early-onset infantile facioscapulohumeral muscular dystrophy: A timely review. Int. J. Mol. Sci. 2020, 21, 7783. Dahlqvist, J.R.; Poulsen, N.S.; Østergaard, S.T.; Fornander, F.; de Stricker Borch, J.; Danielsen, E.R.; Thomsen, C.; Vissing, J. Evaluation of inflammatory lesions over 2 years in facioscapulohumeral muscular dystrophy. Neurology 2002, 95, e1211.
- Steel, D.; Main, M.; Manzur, A.; Muntoni, F.; Munot, P. Clinical features of fascioscapulohumeral muscular dystrophy 1 in childhood. Dev. Med. Child Neurol. 2019, 61, 964–971. Banerji, C.R.S.; Zammit, P.S. Pathomechanisms and biomarkers in facioscapulohumeral muscular dystrophy: Roles of DUX4 and PAX7. EMBO Mol. Med. 2021, 13, e13695.
- Wang, L.H.; Tawil, R. Facioscapulohumeral Dystrophy. Curr. Neurol. Neurosci. Rep. 2016, 16, 66. Lemmers, R.J.; van der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camaño, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; van Ommen, G.J.; Padberg, G.W.; et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010, 329, 1650–1653.
- Dahlqvist, J.R.; Poulsen, N.S.; Østergaard, S.T.; Fornander, F.; de Stricker Borch, J.; Danielsen, E.R.; Thomsen, C.; Vissing, J. Evaluation of inflammatory lesions over 2 years in facioscapulohumeral muscular dystrophy. Neurology 2002, 95, e1211. Lemmers, R.J.L.F.; Tawil, R.; Petek, L.M.; Balog, J.; Block, G.J.; Santen, G.W.; Maell, A.M.; van der Vliet, P.J.; Almomani, R.; Straasheijm, K.R.; et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012, 44, 1370–1374.
- Banerji, C.R.S.; Zammit, P.S. Pathomechanisms and biomarkers in facioscapulohumeral muscular dystrophy: Roles of DUX4 and PAX7. EMBO Mol. Med. 2021, 13, e13695. Hendrickson, P.G.; Doráis, J.A.; Grow, E.J.; Whiddon, J.L.; Lim, J.W.; Wike, C.L.; Weaver, B.D.; Pflueger, C.; Emery, B.R.; Wilcox, A.L.; et al. Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat. Genet. 2017, 49, 925–934.
- Lemmers, R.J.; van der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camaño, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; van Ommen, G.J.; Padberg, G.W.; et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010, 329, 1650–1653. Töhönen, V.; Katayama, S.; Vesterlund, L.; Jouhilahti, E.M.; Sheikhi, M.; Madissoon, E.; Filippini-Cattaneo, G.; Jaconi, M.; Johnsson, A.; Bürglin, T.R.; et al. Novel PRD-like homeodomain transcription factors and retrotransposon elements in early human development. Nat. Commun. 2015, 6, 8207.
- Lemmers, R.J.L.F.; Tawil, R.; Petek, L.M.; Balog, J.; Block, G.J.; Santen, G.W.; Maell, A.M.; van der Vliet, P.J.; Almomani, R.; Straasheijm, K.R.; et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012, 44, 1370–1374. Whiddon, J.L.; Langford, A.T.; Wong, C.J.; Zhong, J.W.; Tapscott, S.J. Conservation and innovation in the DUX4-family gene network. Nat. Genet. 2017, 49, 935–940.
- Hendrickson, P.G.; Doráis, J.A.; Grow, E.J.; Whiddon, J.L.; Lim, J.W.; Wike, C.L.; Weaver, B.D.; Pflueger, C.; Emery, B.R.; Wilcox, A.L.; et al. Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat. Genet. 2017, 49, 925–934. De Iaco, A.; Planet, E.; Coluccio, A.; Verp, S.; Duc, J.; Trono, D. DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat. Genet. 2017, 49, 941–945.
- Töhönen, V.; Katayama, S.; Vesterlund, L.; Jouhilahti, E.M.; Sheikhi, M.; Madissoon, E.; Filippini-Cattaneo, G.; Jaconi, M.; Johnsson, A.; Bürglin, T.R.; et al. Novel PRD-like homeodomain transcription factors and retrotransposon elements in early human development. Nat. Commun. 2015, 6, 8207. Hewitt, J.E. Loss of epigenetic silencing of the DUX4 transcription factor gene in facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 2015, 24, R17–R23.
- Whiddon, J.L.; Langford, A.T.; Wong, C.J.; Zhong, J.W.; Tapscott, S.J. Conservation and innovation in the DUX4-family gene network. Nat. Genet. 2017, 49, 935–940. Snider, L.; Geng, L.N.; Lemmers, R.J.; Kyba, M.; Ware, C.B.; Nelson, A.M.; Tawil, R.; Filippova, G.N.; van der Maarel, S.M.; Tapscott, S.J.; et al. Facioscapulohumeral dystrophy: Incomplete suppression of a retrotransposed gene. PLoS Genet. 2010, 6, e1001181.
- De Iaco, A.; Planet, E.; Coluccio, A.; Verp, S.; Duc, J.; Trono, D. DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat. Genet. 2017, 49, 941–945. Das, S.; Chadwick, B.P. Influence of repressive histone and DNA methylation upon D4Z4 transcription in non-myogenic cells. PLoS ONE 2016, 11, e0160022.
- Hewitt, J.E. Loss of epigenetic silencing of the DUX4 transcription factor gene in facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 2015, 24, R17–R23. Oliva, J.; Galasinski, S.; Richey, A.; Campbell, A.E.; Meyers, M.J.; Modi, N.; Zhong, J.W.; Tawil, R.; Tapscott, S.J.; Sverdrup, F.M. Clinically Advanced p38 Inhibitors Suppress DUX4 Expression in Cellular and Animal Models of Facioscapulohumeral Muscular Dystrophy. J. Pharmacol. Exper. Ther. 2019, 370, 219–230.
- Snider, L.; Geng, L.N.; Lemmers, R.J.; Kyba, M.; Ware, C.B.; Nelson, A.M.; Tawil, R.; Filippova, G.N.; van der Maarel, S.M.; Tapscott, S.J.; et al. Facioscapulohumeral dystrophy: Incomplete suppression of a retrotransposed gene. PLoS Genet. 2010, 6, e1001181. Wijmenga, C.; Hewitt, J.E.; Sandkuijl, L.A.; Clark, L.N.; Wright, T.J.; Dauwerse, H.G.; Gruter, A.M.; Hofker, M.H.; Moerer, P.; Williamson, R. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat. Genet. 1992, 2, 26–30.
- Das, S.; Chadwick, B.P. Influence of repressive histone and DNA methylation upon D4Z4 transcription in non-myogenic cells. PLoS ONE 2016, 11, e0160022. Geng, L.N.; Yao, Z.; Snider, L.; Fong, A.P.; Cech, J.N.; Young, J.M.; van der Maarel, S.M.; Ruzzo, W.L.; Gentleman, R.C.; Tawil, R.; et al. DUX4 activates germline genes, retroelements, and immune mediators: Implications for facioscapulohumeral dystrophy. Dev. Cell 2012, 22, 38–51.
- Oliva, J.; Galasinski, S.; Richey, A.; Campbell, A.E.; Meyers, M.J.; Modi, N.; Zhong, J.W.; Tawil, R.; Tapscott, S.J.; Sverdrup, F.M. Clinically Advanced p38 Inhibitors Suppress DUX4 Expression in Cellular and Animal Models of Facioscapulohumeral Muscular Dystrophy. J. Pharmacol. Exper. Ther. 2019, 370, 219–230. Straub, V.; Bushby, K. The childhood limb-girdle muscular dystrophies. Semin. Pediatr. Neurol. 2006, 13, 104–114.
- Wijmenga, C.; Hewitt, J.E.; Sandkuijl, L.A.; Clark, L.N.; Wright, T.J.; Dauwerse, H.G.; Gruter, A.M.; Hofker, M.H.; Moerer, P.; Williamson, R. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat. Genet. 1992, 2, 26–30. Norwood, F.L.M.; Harling, C.; Chinnery, P.F.; Eagle, M.; Bushby, K.; Straub, V. Prevalence of genetic muscle disease in Northern England: In-depth analysis of a muscle clinic population. Brain 2009, 132, 3175–3186.
- Geng, L.N.; Yao, Z.; Snider, L.; Fong, A.P.; Cech, J.N.; Young, J.M.; van der Maarel, S.M.; Ruzzo, W.L.; Gentleman, R.C.; Tawil, R.; et al. DUX4 activates germline genes, retroelements, and immune mediators: Implications for facioscapulohumeral dystrophy. Dev. Cell 2012, 22, 38–51. Wicklund, M.P. Rare disease clinical trials: Power in numbers. Neurol. Genet. 2016, 2, e92.
- Straub, V.; Bushby, K. The childhood limb-girdle muscular dystrophies. Semin. Pediatr. Neurol. 2006, 13, 104–114. Melacini, P.; Fanin, M.; Duggan, D.J.; Freda, M.P.; Berardinelli, A.; Danieli, G.A.; Barchitta, A.; Hoffman, E.P.; Dalla Volta, S.; Angelini, C. Heart involvement in muscular dystrophy due to sarcoglycan gene mutations. Muscle Nerve 1999, 22, 473–479.
- Norwood, F.L.M.; Harling, C.; Chinnery, P.F.; Eagle, M.; Bushby, K.; Straub, V. Prevalence of genetic muscle disease in Northern England: In-depth analysis of a muscle clinic population. Brain 2009, 132, 3175–3186. Sánchez Riera, C.; Lozanoska-Ochser, B.; Testa, S.; Fornetti, E.; Bouché, M.; Madaro, L. Muscle diversity, heterogeneity, and gradients: Learning from sarcoglycanopathies. Int. J. Mol. Sci. 2021, 22, 2502.
- Wicklund, M.P. Rare disease clinical trials: Power in numbers. Neurol. Genet. 2016, 2, e92. Molina, T.; Fabre, P.; Dumont, N.A. Fibro-adipogenic progenitors in skeletal muscle homeostasis, regeneration and diseases. Open Biol. 2021, 11, 210110.
- Melacini, P.; Fanin, M.; Duggan, D.J.; Freda, M.P.; Berardinelli, A.; Danieli, G.A.; Barchitta, A.; Hoffman, E.P.; Dalla Volta, S.; Angelini, C. Heart involvement in muscular dystrophy due to sarcoglycan gene mutations. Muscle Nerve 1999, 22, 473–479. Kanagawa, M.; Toda, T. Ribitol-phosphate-a newly identified posttranslational glycosylation unit in mammals: Structure, modification enzymes and relationship to human diseases. J. Biochem. 2018, 163, 359–369.
- Sánchez Riera, C.; Lozanoska-Ochser, B.; Testa, S.; Fornetti, E.; Bouché, M.; Madaro, L. Muscle diversity, heterogeneity, and gradients: Learning from sarcoglycanopathies. Int. J. Mol. Sci. 2021, 22, 2502. Brockington, M.; Blake, D.J.; Prandini, P.; Brown, S.C.; Torelli, S.; Benson, M.A.; Ponting, C.P.; Estournet, B.; Romero, N.B.; Mercuri, E. Mutations in the Fukutin-Related Protein Gene (FKRP) Cause a Form of Congenital Muscular Dystrophy with Secondary Laminin a2 Deficiency and Abnormal Glycosylation of a-Dystroglycan. Am. J. Hum. Genet. 2001, 69, 1198–1209.
- Molina, T.; Fabre, P.; Dumont, N.A. Fibro-adipogenic progenitors in skeletal muscle homeostasis, regeneration and diseases. Open Biol. 2021, 11, 210110. Nigro, V.; Savarese, M. Genetic basis of limb-girdle muscular dystrophies: The 2014 update. Acta Myol. 2014, 33, 1–12.
- Kanagawa, M.; Toda, T. Ribitol-phosphate-a newly identified posttranslational glycosylation unit in mammals: Structure, modification enzymes and relationship to human diseases. J. Biochem. 2018, 163, 359–369. Thompson, R.; Straub, V. Limb-girdle muscular dystrophies—International collaborations for translational research. Nat. Rev. Neurol. 2016, 12, 294–309.
- Brockington, M.; Blake, D.J.; Prandini, P.; Brown, S.C.; Torelli, S.; Benson, M.A.; Ponting, C.P.; Estournet, B.; Romero, N.B.; Mercuri, E. Mutations in the Fukutin-Related Protein Gene (FKRP) Cause a Form of Congenital Muscular Dystrophy with Secondary Laminin a2 Deficiency and Abnormal Glycosylation of a-Dystroglycan. Am. J. Hum. Genet. 2001, 69, 1198–1209. Wu, B.; Shah, S.N.; Lu, P.; Richardson, S.M.; Bollinger, L.E.; Blaeser, A.; Madden, K.L.; Sun, Y.; Luckie, T.M.; Cox, M.D. Glucocorticoid steroid and alendronate treatment alleviates dystrophic phenotype with enhanced functional glycosylation of α-dystroglycan in mouse model of limb-girdle muscular dystrophy with FKRPP448L mutation. Am. J. Pathol. 2016, 186, 1635–1648.
- Nigro, V.; Savarese, M. Genetic basis of limb-girdle muscular dystrophies: The 2014 update. Acta Myol. 2014, 33, 1–12. Taniguchi, K.; Kobayashi, K.; Saito, K.; Yamanouchi, H.; Ohnuma, A.; Hayashi, Y.K.; Manya, H.; Jin, D.K.; Lee, M.; Parano, E. Worldwide distribution and broader clinical spectrum of muscle-eye-brain disease. Hum. Mol. Genet. 2003, 12, 527–534.
- Thompson, R.; Straub, V. Limb-girdle muscular dystrophies—International collaborations for translational research. Nat. Rev. Neurol. 2016, 12, 294–309. de Bernabé, D.B.V.; Voit, T.; Longman, C.; Steinbrecher, A.; Straub, V.; Yuva, Y.; Herrmann, R.; Sperner, J.; Korenke, C.; Diesen, C.; et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J. Med. Genet. 2004, 41, e61.
- Wu, B.; Shah, S.N.; Lu, P.; Richardson, S.M.; Bollinger, L.E.; Blaeser, A.; Madden, K.L.; Sun, Y.; Luckie, T.M.; Cox, M.D. Glucocorticoid steroid and alendronate treatment alleviates dystrophic phenotype with enhanced functional glycosylation of α-dystroglycan in mouse model of limb-girdle muscular dystrophy with FKRPP448L mutation. Am. J. Pathol. 2016, 186, 1635–1648. Chan, Y.M.; Keramaris-Vrantsis, E.; Lidov, H.G.; Norton, J.H.; Zinchenko, N.; Gruber, H.E.; Thresher, R.; Blake, D.J.; Ashar, J.; Rosenfeld, J.; et al. Fukutin-related protein is essential for mouse muscle, brain and eye development and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum. Mol. Genet. 2010, 19, 3995–4006.
- Taniguchi, K.; Kobayashi, K.; Saito, K.; Yamanouchi, H.; Ohnuma, A.; Hayashi, Y.K.; Manya, H.; Jin, D.K.; Lee, M.; Parano, E. Worldwide distribution and broader clinical spectrum of muscle-eye-brain disease. Hum. Mol. Genet. 2003, 12, 527–534. Brancaccio, A. A molecular overview of the primary dystroglycanopathies. J. Cell. Mol. Med. 2018, 23, 3058–3062.
- de Bernabé, D.B.V.; Voit, T.; Longman, C.; Steinbrecher, A.; Straub, V.; Yuva, Y.; Herrmann, R.; Sperner, J.; Korenke, C.; Diesen, C.; et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J. Med. Genet. 2004, 41, e61. Wu, B.; Shah, S.N.; Lu, P.; Bollinger, L.E.; Blaeser, A.; Sparks, S.; Harper, A.D.; Lu, Q.L. Long-term treatment of tamoxifen and raloxifene alleviates dystrophic phenotype and enhances muscle functions of FKRP dystroglycanopathy. Am. J. Pathol. 2018, 188, 1069–1080.
- Chan, Y.M.; Keramaris-Vrantsis, E.; Lidov, H.G.; Norton, J.H.; Zinchenko, N.; Gruber, H.E.; Thresher, R.; Blake, D.J.; Ashar, J.; Rosenfeld, J.; et al. Fukutin-related protein is essential for mouse muscle, brain and eye development and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum. Mol. Genet. 2010, 19, 3995–4006. Gui, Y.S.; Wang, L.; Tian, X.; Li, X.; Ma, A.; Zhou, W.; Zeng, N.; Zhang, J.; Cai, B.; Zhang, H. mTOR overactivation and compromised autophagy in the pathogenesis of pulmonary fibrosis. PLoS ONE 2015, 10, e0138625.
- Brancaccio, A. A molecular overview of the primary dystroglycanopathies. J. Cell. Mol. Med. 2018, 23, 3058–3062. Prandini, P.; Berardinelli, A.; Fanin, M.; Morello, F.; Zardini, E.; Pichiecchio, A.; Uggetti, C.; Lanzi, G.; Angelini, C.; Pegoraro, E. LAMA2 loss-of-function mutation in a girl with a mild congenital muscular dystrophy. Neurology 2004, 63, 1118–1121.
- Wu, B.; Shah, S.N.; Lu, P.; Bollinger, L.E.; Blaeser, A.; Sparks, S.; Harper, A.D.; Lu, Q.L. Long-term treatment of tamoxifen and raloxifene alleviates dystrophic phenotype and enhances muscle functions of FKRP dystroglycanopathy. Am. J. Pathol. 2018, 188, 1069–1080. Geranmayeh, F.; Clement, E.; Feng, L.H.; Sewry, C.; Pagan, J.; Mein, R.; Abbs, S.; Brueton, L.; Childs, A.M.; Jungbluth, H.; et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul. Disord. 2010, 20, 241–250.
- Gui, Y.S.; Wang, L.; Tian, X.; Li, X.; Ma, A.; Zhou, W.; Zeng, N.; Zhang, J.; Cai, B.; Zhang, H. mTOR overactivation and compromised autophagy in the pathogenesis of pulmonary fibrosis. PLoS ONE 2015, 10, e0138625. Leite, C.C.; Lucato, L.T.; Martin, M.G.; Ferreira, L.G.; Resende, M.B.; Carvalho, M.S.; Marie, S.K.; Jinkins, J.R.; Reed, U.C. Merosin-deficient congenital muscular dystrophy (CMD): A study of 25 Brazilian patients using MRI. Ped. Radiol. 2005, 35, 572–579.
- Prandini, P.; Berardinelli, A.; Fanin, M.; Morello, F.; Zardini, E.; Pichiecchio, A.; Uggetti, C.; Lanzi, G.; Angelini, C.; Pegoraro, E. LAMA2 loss-of-function mutation in a girl with a mild congenital muscular dystrophy. Neurology 2004, 63, 1118–1121. Bönnemann, C.G.; Wang, C.H.; Quijano-Roy, S.; Deconinck, N.; Bertini, E.; Ferreiro, A.; Muntoni, F.; Sewry, C.; Béroud, C.; Mathews, K.D.; et al. Members of International Standard of Care Committee for Congenital Muscular Dystrophies. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul. Disord. 2014, 24, 289–311.
- Geranmayeh, F.; Clement, E.; Feng, L.H.; Sewry, C.; Pagan, J.; Mein, R.; Abbs, S.; Brueton, L.; Childs, A.M.; Jungbluth, H.; et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul. Disord. 2010, 20, 241–250. Arreguin, A.J.; Colognato, H. Brain dysfunction in Lama2-Related congenital muscular dystrophy: Lessons from human case reports and mouse models. Front. Mol. Neurosci. 2020, 13, 118.
- Leite, C.C.; Lucato, L.T.; Martin, M.G.; Ferreira, L.G.; Resende, M.B.; Carvalho, M.S.; Marie, S.K.; Jinkins, J.R.; Reed, U.C. Merosin-deficient congenital muscular dystrophy (CMD): A study of 25 Brazilian patients using MRI. Ped. Radiol. 2005, 35, 572–579. Xiong, H.; Tan, D.; Wang, S.; Song, S.; Yang, H.; Gao, K.; Liu, A.; Jiao, H.; Mao, B.; Ding, J. Genotype/phenotype analysis in Chinese laminin-α2 deficient congenital muscular dystrophy patients. Clin. Genet. 2015, 87, 233–243.
- Bönnemann, C.G.; Wang, C.H.; Quijano-Roy, S.; Deconinck, N.; Bertini, E.; Ferreiro, A.; Muntoni, F.; Sewry, C.; Béroud, C.; Mathews, K.D.; et al. Members of International Standard of Care Committee for Congenital Muscular Dystrophies. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul. Disord. 2014, 24, 289–311. Sarkozy, A.; Foley, A.R.; Zambon, A.A.; Bönnemann, C.G.; Muntoni, F. LAMA2-Related Dystrophies: Clinical Phenotypes, Disease Biomarkers, and Clinical Trial Readiness. Front. Mol. Neurosci. 2020, 13, 123.
- Arreguin, A.J.; Colognato, H. Brain dysfunction in Lama2-Related congenital muscular dystrophy: Lessons from human case reports and mouse models. Front. Mol. Neurosci. 2020, 13, 118. Taniguchi, M.; Kurahashi, H.; Noguchi, S.; Sese, J.; Okinaga, T.; Tsukahara, T.; Guicheney, P.; Ozono, K.; Nishino, I.; Morishita, S.; et al. Expression profiling of muscles from Fukuyama-type congenital muscular dystrophy and laminin-alpha 2 deficient congenital muscular dystrophy; is congenital muscular dystrophy a primary fibrotic disease? Biochem. Biophys. Res. Commun. 2006, 342, 489–502.
- Xiong, H.; Tan, D.; Wang, S.; Song, S.; Yang, H.; Gao, K.; Liu, A.; Jiao, H.; Mao, B.; Ding, J. Genotype/phenotype analysis in Chinese laminin-α2 deficient congenital muscular dystrophy patients. Clin. Genet. 2015, 87, 233–243. Zhu, S.; Zhang, Q.; Gudise, C.; Wei, L.; Smith, E.; Zeng, Y. Synthesis and biological evaluation of febrifugine analogues as potential antimalarial agents. Bioorg. Med. Chem. 2009, 17, 4496–4502.
- Sarkozy, A.; Foley, A.R.; Zambon, A.A.; Bönnemann, C.G.; Muntoni, F. LAMA2-Related Dystrophies: Clinical Phenotypes, Disease Biomarkers, and Clinical Trial Readiness. Front. Mol. Neurosci. 2020, 13, 123. Elbaz, M.; Yanay, N.; Aga-Mizrachi, S.; Brunschwig, Z.; Kassis, I.; Ettinger, K.; Barak, V.; Nevo, Y. Losartan, a therapeutic candidate in congenital muscular dystrophy: Studies in the dy(2J)/dy(2J) mouse. Ann. Neurol. 2012, 71, 699–708.
- Taniguchi, M.; Kurahashi, H.; Noguchi, S.; Sese, J.; Okinaga, T.; Tsukahara, T.; Guicheney, P.; Ozono, K.; Nishino, I.; Morishita, S.; et al. Expression profiling of muscles from Fukuyama-type congenital muscular dystrophy and laminin-alpha 2 deficient congenital muscular dystrophy; is congenital muscular dystrophy a primary fibrotic disease? Biochem. Biophys. Res. Commun. 2006, 342, 489–502. Meinen, S.; Lin, S.; Ruegg, M.A. Angiotensin II type 1 receptor antagonists alleviate muscle pathology in the mouse model for laminin-α2-deficient congenital muscular dystrophy (MDC1A). Skelet. Muscle 2012, 2, 18.
- Zhu, S.; Zhang, Q.; Gudise, C.; Wei, L.; Smith, E.; Zeng, Y. Synthesis and biological evaluation of febrifugine analogues as potential antimalarial agents. Bioorg. Med. Chem. 2009, 17, 4496–4502. Accorsi, A.; Mehuron, T.; Kumar, A.; Rhee, Y.; Girgenrath, M. Integrin dysregulation as a possible driver of matrix remodeling in Laminin-deficient congenital muscular dystrophy (MDC1A). J. Neuromuscul. Dis. 2015, 2, 51–61.
- Elbaz, M.; Yanay, N.; Aga-Mizrachi, S.; Brunschwig, Z.; Kassis, I.; Ettinger, K.; Barak, V.; Nevo, Y. Losartan, a therapeutic candidate in congenital muscular dystrophy: Studies in the dy(2J)/dy(2J) mouse. Ann. Neurol. 2012, 71, 699–708. Accorsi, A.; Kumar, A.; Rhee, Y.; Miller, A.; Girgenrath, M. IGF-1/GH axis enhances losartan treatment in Lama2-related muscular dystrophy. Hum. Mol. Genet. 2016, 25, 4624–4634.
- Meinen, S.; Lin, S.; Ruegg, M.A. Angiotensin II type 1 receptor antagonists alleviate muscle pathology in the mouse model for laminin-α2-deficient congenital muscular dystrophy (MDC1A). Skelet. Muscle 2012, 2, 18. Oliveira-Santos, A.; Dagda, M.; Wittmann, J.; Smalley, R.; Burkin, D.J. Vemurafenib improves muscle histopathology in a mouse model of LAMA2-related congenital muscular dystrophy. Dis. Model. Mech. 2023, 16, dmm049916.
- Accorsi, A.; Mehuron, T.; Kumar, A.; Rhee, Y.; Girgenrath, M. Integrin dysregulation as a possible driver of matrix remodeling in Laminin-deficient congenital muscular dystrophy (MDC1A). J. Neuromuscul. Dis. 2015, 2, 51–61. Erb, M.; Meinen, S.; Barzaghi, P.; Sumanovski, L.T.; Courdier-Früh, I.; Rüegg, M.A.; Meier, T. Omigapil Ameliorates the Pathology of Muscle Dystrophy Caused by Laminin-α2 Deficiency. J. Pharm. Exp. Ther. 2009, 331, 787–795.
- Accorsi, A.; Kumar, A.; Rhee, Y.; Miller, A.; Girgenrath, M. IGF-1/GH axis enhances losartan treatment in Lama2-related muscular dystrophy. Hum. Mol. Genet. 2016, 25, 4624–4634. Yamauchi, J.; Kumar, A.; Duarte, L.; Mehuron, T.; Girgenrath, M. Triggering regeneration and tackling apoptosis: A combinatorial approach to treating congenital muscular dystrophy type 1A. Hum. Mol. Genet. 2013, 22, 4306–4317.
- Oliveira-Santos, A.; Dagda, M.; Wittmann, J.; Smalley, R.; Burkin, D.J. Vemurafenib improves muscle histopathology in a mouse model of LAMA2-related congenital muscular dystrophy. Dis. Model. Mech. 2023, 16, dmm049916. Carmignac, V.; Quere, R.; Durbeej, M. Proteasome inhibition improves the muscle of laminin alpha2 chain deficient mice. Hum. Mol. Genet. 2011, 20, 541–552.
- Erb, M.; Meinen, S.; Barzaghi, P.; Sumanovski, L.T.; Courdier-Früh, I.; Rüegg, M.A.; Meier, T. Omigapil Ameliorates the Pathology of Muscle Dystrophy Caused by Laminin-α2 Deficiency. J. Pharm. Exp. Ther. 2009, 331, 787–795. Carmignac, V.; Svensson, M.; Korner, Z.; Elowsson, L.; Matsumura, C.; Gawlik, K.I.; Allamand, V.; Durbeej, M. Autophagy is increased in laminin alpha2 chain deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum. Mol. Genet. 2011, 20, 4891–4902.
- Yamauchi, J.; Kumar, A.; Duarte, L.; Mehuron, T.; Girgenrath, M. Triggering regeneration and tackling apoptosis: A combinatorial approach to treating congenital muscular dystrophy type 1A. Hum. Mol. Genet. 2013, 22, 4306–4317. Körner, Z.; Fontes-Oliveira, C.C.; Holmberg, J.; Carmignac, V.; Durbeej, M. Bortezomib Partially Improves Laminin α2 Chain–Deficient Muscular Dystrophy. Am. J. Path. 2014, 184, 1518–1528.
- Carmignac, V.; Quere, R.; Durbeej, M. Proteasome inhibition improves the muscle of laminin alpha2 chain deficient mice. Hum. Mol. Genet. 2011, 20, 541–552. Lamandé, S.R.; Bateman, J.F. Collagen VI disorders: Insights on form and function in the extracellular matrix and beyond. Matrix Biol. 2017, 71–72, 348–367.
- Carmignac, V.; Svensson, M.; Korner, Z.; Elowsson, L.; Matsumura, C.; Gawlik, K.I.; Allamand, V.; Durbeej, M. Autophagy is increased in laminin alpha2 chain deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum. Mol. Genet. 2011, 20, 4891–4902. Pfaff, M.; Aumailley, M.; Specks, U.; Knolle, J.; Zerwes, H.G.; Timpl, R. Integrin and Arg-Gly-Asp dependence of cell adhesion to the native and unfolded triple helix of collagen type VI. Exp. Cell Res. 1993, 206, 167–176.
- Körner, Z.; Fontes-Oliveira, C.C.; Holmberg, J.; Carmignac, V.; Durbeej, M. Bortezomib Partially Improves Laminin α2 Chain–Deficient Muscular Dystrophy. Am. J. Path. 2014, 184, 1518–1528. Nanda, A.; Carson-Walter, E.B.; Seaman, S.; Barber, T.D.; Stampfl, J.; Singh, S.; Vogelstein, B.; Kinzler, K.W.; St Croix, B. TEM8 interacts with the cleaved C5 domain of collagen alpha 3(VI). Cancer Res. 2004, 64, 817–820.
- Lamandé, S.R.; Bateman, J.F. Collagen VI disorders: Insights on form and function in the extracellular matrix and beyond. Matrix Biol. 2017, 71–72, 348–367. Buraschi, S.; Neill, T.; Iozzo, R.V. Decorin is a devouring proteoglycan: Remodeling of intracellular catabolism via autophagy and mitophagy. Matrix Biol. 2019, 75–76, 260–270.
- Pfaff, M.; Aumailley, M.; Specks, U.; Knolle, J.; Zerwes, H.G.; Timpl, R. Integrin and Arg-Gly-Asp dependence of cell adhesion to the native and unfolded triple helix of collagen type VI. Exp. Cell Res. 1993, 206, 167–176. Noguchi, S.; Ogawa, M.; Malicdan, M.C.; Nonaka, I.; Nishino, I. Muscle weakness and fibrosis due to cell autonomous and non-cell autonomous events in Collagen VI deficient congenital muscular dystrophy. EBioMedicine 2017, 15, 193–202.
- Nanda, A.; Carson-Walter, E.B.; Seaman, S.; Barber, T.D.; Stampfl, J.; Singh, S.; Vogelstein, B.; Kinzler, K.W.; St Croix, B. TEM8 interacts with the cleaved C5 domain of collagen alpha 3(VI). Cancer Res. 2004, 64, 817–820. Vita, G.L.; Sframeli, M.; Licata, N.; Bitto, A.; Romeo, S.; Frisone, F.; Ciranni, A.; Pallio, G.; Mannino, F.; Aguennouz, M.; et al. A Phase 1/2 Study of Flavocoxid, an Oral NF-κB Inhibitor, in Duchenne Muscular Dystrophy. Brain Sci. 2021, 11, 115.
- Buraschi, S.; Neill, T.; Iozzo, R.V. Decorin is a devouring proteoglycan: Remodeling of intracellular catabolism via autophagy and mitophagy. Matrix Biol. 2019, 75–76, 260–270. Raffaghello, L.; Principi, E.; Baratto, S.; Panicucci, C.; Pintus, S.; Antonini, F.; Del Zotto, G.; Benzi, A.; Bruzzone, S.; Scudieri, P.; et al. P2X7 Receptor Antagonist Reduces Fibrosis and Inflammation in a Mouse Model of Alpha-Sarcoglycan Muscular Dystrophy. Pharmaceuticals 2022, 15, 89.
- Noguchi, S.; Ogawa, M.; Malicdan, M.C.; Nonaka, I.; Nishino, I. Muscle weakness and fibrosis due to cell autonomous and non-cell autonomous events in Collagen VI deficient congenital muscular dystrophy. EBioMedicine 2017, 15, 193–202. Sreetama, S.C.; Chandra, G.; Meulen, J.H.V.; Ahmad, M.M.; Suzuki, P.; Bhuvanendran, S.; Nagaraju, K.; Hoffman, E.P.; Jaiswal, J.K. Membrane Stabilization by Modified Steroid Offers a Potential Therapy for Muscular Dystrophy Due to Dysferlin Deficit. Mol. Ther. 2018, 26, 2231–2242.
- Vita, G.L.; Sframeli, M.; Licata, N.; Bitto, A.; Romeo, S.; Frisone, F.; Ciranni, A.; Pallio, G.; Mannino, F.; Aguennouz, M.; et al. A Phase 1/2 Study of Flavocoxid, an Oral NF-κB Inhibitor, in Duchenne Muscular Dystrophy. Brain Sci. 2021, 11, 115. Kang, J.; Feng, D.; Yang, F.; Tian, X.; Han, W.; Jia, H. Comparison of rapamycin and methylprednisolone for treating inflammatory muscle disease in a murine model of experimental autoimmune myositis. Exp. Ther. Med. 2020, 20, 219–226.
- Raffaghello, L.; Principi, E.; Baratto, S.; Panicucci, C.; Pintus, S.; Antonini, F.; Del Zotto, G.; Benzi, A.; Bruzzone, S.; Scudieri, P.; et al. P2X7 Receptor Antagonist Reduces Fibrosis and Inflammation in a Mouse Model of Alpha-Sarcoglycan Muscular Dystrophy. Pharmaceuticals 2022, 15, 89. Saxton, R.A.; Sabatini, D.M. mTor signaling in growth, metabolism, and disease. Cell 2017, 169, 361–371.
- Sreetama, S.C.; Chandra, G.; Meulen, J.H.V.; Ahmad, M.M.; Suzuki, P.; Bhuvanendran, S.; Nagaraju, K.; Hoffman, E.P.; Jaiswal, J.K. Membrane Stabilization by Modified Steroid Offers a Potential Therapy for Muscular Dystrophy Due to Dysferlin Deficit. Mol. Ther. 2018, 26, 2231–2242. Zhang, P.; Liang, X.; Shan, T.; Jiang, Q.; Deng, C.; Zheng, R.; Kuang, S. mTOR is necessary for proper satellite cell activity and skeletal muscle regeneration. Biochem. Biophys. Res. Commun. 2015, 463, 102–108.
- Kang, J.; Feng, D.; Yang, F.; Tian, X.; Han, W.; Jia, H. Comparison of rapamycin and methylprednisolone for treating inflammatory muscle disease in a murine model of experimental autoimmune myositis. Exp. Ther. Med. 2020, 20, 219–226. Foltz, S.J.; Luan, J.; Call, J.A.; Patel, A.; Peissig, K.B.; Fortunato, M.J.; Beedle, A.M. Four-week rapamycin treatment improves muscular dystrophy in a fukutin-deficient mouse model of dystroglycanopathy. Skelet. Muscle 2016, 6, 20.
- Saxton, R.A.; Sabatini, D.M. mTor signaling in growth, metabolism, and disease. Cell 2017, 169, 361–371. Andre, A.B.; Zhang, L.; Nix, J.D.; Elmadbouly, N.; Lucas, A.R.; Wilson-Rawls, J.; Rawls, A. Myxomavirus Serp-1 Protein Ameliorates Inflammation in a Mouse Model of Duchenne Muscular Dystrophy. Biomedicines 2022, 10, 1154.
- Zhang, P.; Liang, X.; Shan, T.; Jiang, Q.; Deng, C.; Zheng, R.; Kuang, S. mTOR is necessary for proper satellite cell activity and skeletal muscle regeneration. Biochem. Biophys. Res. Commun. 2015, 463, 102–108. Lluís, F.; Roma, J.; Suelves, M.; Parra, M.; Aniorte, G.; Gallardo, E.; Illa, I.; Rodríguez, L.; Hughes, S.M.; Carmeliet, P.; et al. Urokinase-Dependent Plasminogen Activation Is Required for Efficient Skeletal Muscle Regeneration In Vivo. Blood 2001, 97, 1703–1711.
- Foltz, S.J.; Luan, J.; Call, J.A.; Patel, A.; Peissig, K.B.; Fortunato, M.J.; Beedle, A.M. Four-week rapamycin treatment improves muscular dystrophy in a fukutin-deficient mouse model of dystroglycanopathy. Skelet. Muscle 2016, 6, 20. Ardite, E.; Perdiguero, E.; Vidal, B.; Gutarra, S.; Serrano, A.L.; Muñoz-Cánoves, P. PAI-1–Regulated MiR-21 Defines a Novel Age-Associated Fibrogenic Pathway in Muscular Dystrophy. J. Cell Biol. 2012, 196, 163–175.
- Andre, A.B.; Zhang, L.; Nix, J.D.; Elmadbouly, N.; Lucas, A.R.; Wilson-Rawls, J.; Rawls, A. Myxomavirus Serp-1 Protein Ameliorates Inflammation in a Mouse Model of Duchenne Muscular Dystrophy. Biomedicines 2022, 10, 1154.
- Lluís, F.; Roma, J.; Suelves, M.; Parra, M.; Aniorte, G.; Gallardo, E.; Illa, I.; Rodríguez, L.; Hughes, S.M.; Carmeliet, P.; et al. Urokinase-Dependent Plasminogen Activation Is Required for Efficient Skeletal Muscle Regeneration In Vivo. Blood 2001, 97, 1703–1711.
- Ardite, E.; Perdiguero, E.; Vidal, B.; Gutarra, S.; Serrano, A.L.; Muñoz-Cánoves, P. PAI-1–Regulated MiR-21 Defines a Novel Age-Associated Fibrogenic Pathway in Muscular Dystrophy. J. Cell Biol. 2012, 196, 163–175.