In the early stages of Alzheimer–Perusini’s disease (AD), individuals often experience vision-related issues such as color vision impairment, reduced contrast sensitivity, and visual acuity problems. As the disease progresses, there is a connection with glaucoma and age-related macular degeneration (AMD) leading to retinal cell death. The retina’s involvement suggests a link with the hippocampus, where most AD forms start. A thinning of the retinal nerve fiber layer (RNFL) due to the loss of retinal ganglion cells (RGCs) is seen as a potential AD diagnostic marker using electroretinography (ERG) and optical coherence tomography (OCT). Amyloid beta fragments (Aβ), found in the eye’s vitreous and aqueous humor, are also present in the cerebrospinal fluid (CSF) and accumulate in the retina. Aβ is known to cause tau hyperphosphorylation, leading to its buildup in various retinal layers.
- eye
- brain
- retinal dystrophies
- Alzheimer–Perusini
1. Retinal Degeneration in Alzheimer’s Disease (AD)
2. Histopathological Alterations in Different Eye Cytotypes Due to β-Amyloid Production
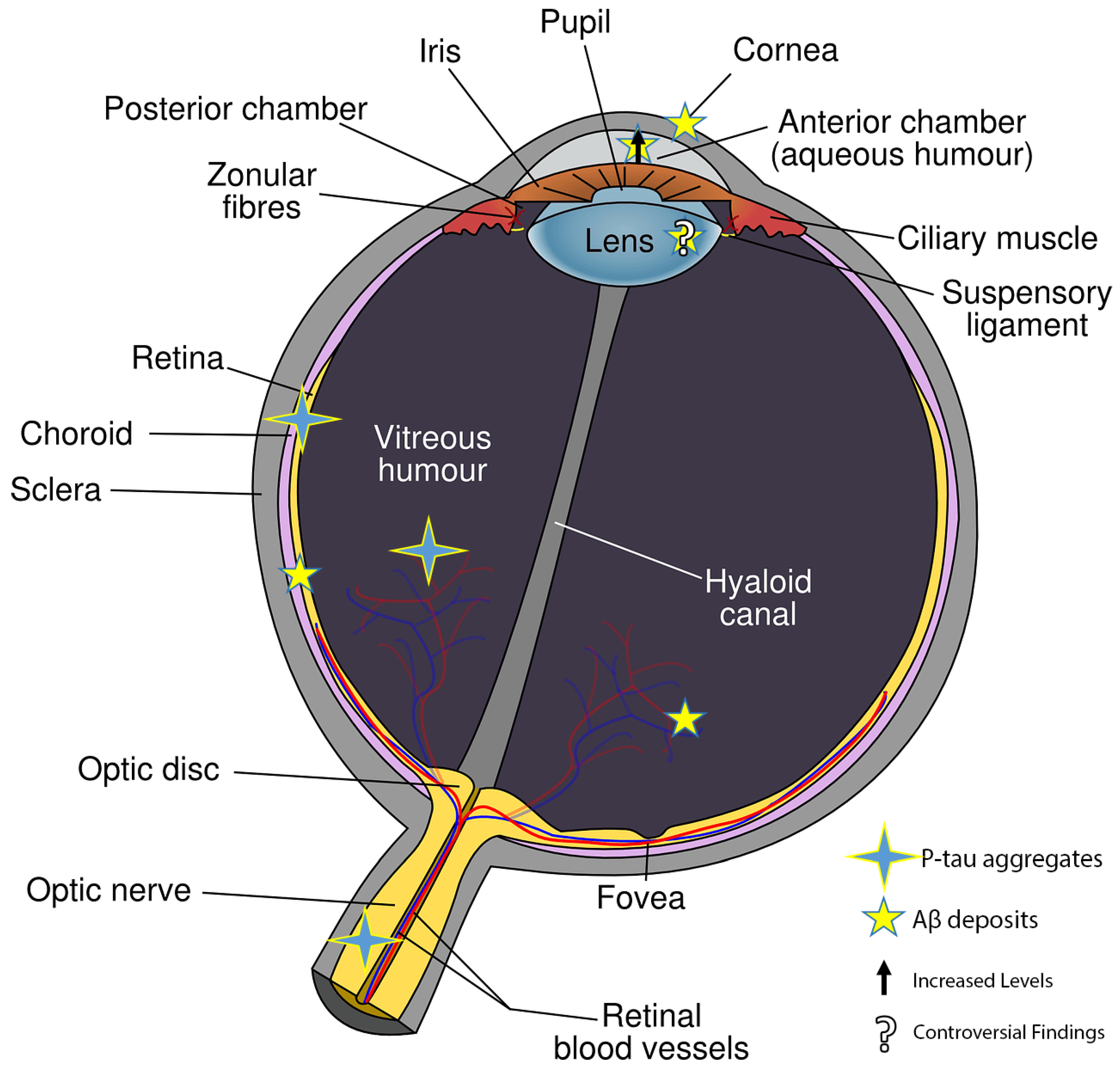
3. Retinal Histopathological Abnormalities in AD Mouse Models
4. The Role of the Prion Protein in Retinal Allostasis
References
- Moons, L.; De Groef, L. Multimodal retinal imaging to detect and understand Alzheimer’s and Parkinson’s disease. Curr. Opin. Neurobiol. 2022, 72, 1–7.
- Shi, H.; Koronyo, Y.; Rentsendorj, A.; Fuchs, D.T.; Sheyn, J.; Black, K.L.; Mirzaei, N.; Koronyo-Hamaoui, M. Retinal Vasculopathy in Alzheimer’s Disease. Front. Neurosci. 2021, 15, 731614.
- Yoon, S.P.; Grewal, D.S.; Thompson, A.C.; Polascik, B.W.; Dunn, C.; Burke, J.R.; Fekrat, S. Retinal Microvascular and Neurodegenerative Changes in Alzheimer’s Disease and Mild Cognitive Impairment Compared with Control Participants. Ophthalmol. Retin. 2019, 3, 489–499.
- Ashok, A.; Singh, N.; Chaudhary, S.; Bellamkonda, V.; Kritikos, A.E.; Wise, A.S.; Rana, N.; McDonald, D.; Ayyagari, R. Retinal Degeneration and Alzheimer’s Disease: An Evolving Link. Int. J. Mol. Sci. 2020, 21, 7290.
- Asanad, S.; Ross-Cisneros, F.N.; Nassisi, M.; Barron, E.; Karanjia, R.; Sadun, A.A. The Retina in Alzheimer’s Disease: Histomorphometric Analysis of an Ophthalmologic Biomarker. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1491–1500.
- Gupta, V.; Gupta, V.B.; Chitranshi, N.; Gangoda, S.; Vander Wall, R.; Abbasi, M.; Golzan, M.; Dheer, Y.; Shah, T.; Avolio, A.; et al. One protein, multiple pathologies: Multifaceted involvement of amyloid beta in neurodegenerative disorders of the brain and retina. Cell Mol. Life Sci. 2016, 73, 4279–4297.
- Ukalovic, K.; Cao, S.; Lee, S.; Tang, Q.; Beg, M.F.; Sarunic, M.V.; Hsiung, G.R.; Mackenzie, I.R.; Hirsch-Reinshagen, V.; Cui, J.Z.; et al. Drusen in the Peripheral Retina of the Alzheimer’s Eye. Curr. Alzheimer Res. 2018, 15, 743–750.
- Blanks, J.C.; Schmidt, S.Y.; Torigoe, Y.; Porrello, K.V.; Hinton, D.R.; Blanks, R.H. Retinal pathology in Alzheimer’s disease. II. Regional neuron loss and glial changes in GCL. Neurobiol. Aging 1996, 17, 385–395.
- Guo, L.Y.; Alekseev, O.; Li, Y.; Song, Y.; Dunaief, J.L. Iron increases APP translation and amyloid-beta production in the retina. Exp. Eye Res. 2014, 129, 31–37.
- Ohno-Matsui, K. Parallel findings in age-related macular degeneration and Alzheimer’s disease. Prog. Retin. Eye Res. 2011, 30, 217–238.
- Ong, S.S.; Proia, A.D.; Whitson, H.E.; Farsiu, S.; Doraiswamy, P.M.; Lad, E.M. Ocular amyloid imaging at the crossroad of Alzheimer’s disease and age-related macular degeneration: Implications for diagnosis and therapy. J. Neurol. 2019, 266, 1566–1577.
- Donato, L.; Morda, D.; Scimone, C.; Alibrandi, S.; D’Angelo, R.; Sidoti, A. How Many Alzheimer-Perusini’s Atypical Forms Do We Still Have to Discover? Biomedicines 2023, 11, 2035.
- den Haan, J.; Morrema, T.H.J.; Verbraak, F.D.; de Boer, J.F.; Scheltens, P.; Rozemuller, A.J.; Bergen, A.A.B.; Bouwman, F.H.; Hoozemans, J.J. Amyloid-beta and phosphorylated tau in post-mortem Alzheimer’s disease retinas. Acta Neuropathol. Commun. 2018, 6, 147.
- Koronyo, Y.; Biggs, D.; Barron, E.; Boyer, D.S.; Pearlman, J.A.; Au, W.J.; Kile, S.J.; Blanco, A.; Fuchs, D.T.; Ashfaq, A.; et al. Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer’s disease. JCI Insight 2017, 2, e93621.
- Shah, T.M.; Gupta, S.M.; Chatterjee, P.; Campbell, M.; Martins, R.N. Beta-amyloid sequelae in the eye: A critical review on its diagnostic significance and clinical relevance in Alzheimer’s disease. Mol. Psychiatry 2017, 22, 353–363.
- Cerquera-Jaramillo, M.A.; Nava-Mesa, M.O.; Gonzalez-Reyes, R.E.; Tellez-Conti, C.; de-la-Torre, A. Visual Features in Alzheimer’s Disease: From Basic Mechanisms to Clinical Overview. Neural Plast. 2018, 2018, 2941783.
- Berisha, F.; Feke, G.T.; Trempe, C.L.; McMeel, J.W.; Schepens, C.L. Retinal abnormalities in early Alzheimer’s disease. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2285–2289.
- Liu, D.; Zhang, L.; Li, Z.; Zhang, X.; Wu, Y.; Yang, H.; Min, B.; Zhang, X.; Ma, D.; Lu, Y. Thinner changes of the retinal nerve fiber layer in patients with mild cognitive impairment and Alzheimer’s disease. BMC Neurol. 2015, 15, 14.
- Marziani, E.; Pomati, S.; Ramolfo, P.; Cigada, M.; Giani, A.; Mariani, C.; Staurenghi, G. Evaluation of retinal nerve fiber layer and ganglion cell layer thickness in Alzheimer’s disease using spectral-domain optical coherence tomography. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5953–5958.
- Polo, V.; Rodrigo, M.J.; Garcia-Martin, E.; Otin, S.; Larrosa, J.M.; Fuertes, M.I.; Bambo, M.P.; Pablo, L.E.; Satue, M. Visual dysfunction and its correlation with retinal changes in patients with Alzheimer’s disease. Eye 2017, 31, 1034–1041.
- Jones-Odeh, E.; Hammond, C.J. How strong is the relationship between glaucoma, the retinal nerve fibre layer, and neurodegenerative diseases such as Alzheimer’s disease and multiple sclerosis? Eye 2015, 29, 1270–1284.
- Sivak, J.M. The aging eye: Common degenerative mechanisms between the Alzheimer’s brain and retinal disease. Investig. Ophthalmol. Vis. Sci. 2013, 54, 871–880.
- Liao, H.; Zhu, Z.; Peng, Y. Potential Utility of Retinal Imaging for Alzheimer’s Disease: A Review. Front. Aging Neurosci. 2018, 10, 188.
- Ngolab, J.; Honma, P.; Rissman, R.A. Reflections on the Utility of the Retina as a Biomarker for Alzheimer’s Disease: A Literature Review. Neurol. Ther. 2019, 8, 57–72.
- Tsokolas, G.; Tsaousis, K.T.; Diakonis, V.F.; Matsou, A.; Tyradellis, S. Optical Coherence Tomography Angiography in Neurodegenerative Diseases: A Review. Eye Brain 2020, 12, 73–87.
- Ratnayaka, J.A.; Serpell, L.C.; Lotery, A.J. Dementia of the eye: The role of amyloid beta in retinal degeneration. Eye 2015, 29, 1013–1026.
- Liu, B.; Rasool, S.; Yang, Z.; Glabe, C.G.; Schreiber, S.S.; Ge, J.; Tan, Z. Amyloid-peptide vaccinations reduce beta-amyloid plaques but exacerbate vascular deposition and inflammation in the retina of Alzheimer’s transgenic mice. Am. J. Pathol. 2009, 175, 2099–2110.
- Liu, C.; Zhang, C.W.; Zhou, Y.; Wong, W.Q.; Lee, L.C.; Ong, W.Y.; Yoon, S.O.; Hong, W.; Fu, X.Y.; Soong, T.W.; et al. APP upregulation contributes to retinal ganglion cell degeneration via JNK3. Cell Death Differ. 2018, 25, 663–678.
- Zhao, Y.; Bhattacharjee, S.; Jones, B.M.; Hill, J.M.; Clement, C.; Sambamurti, K.; Dua, P.; Lukiw, W.J. Beta-Amyloid Precursor Protein (betaAPP) Processing in Alzheimer’s Disease (AD) and Age-Related Macular Degeneration (AMD). Mol. Neurobiol. 2015, 52, 533–544.
- Prasad, T.; Zhu, P.; Verma, A.; Chakrabarty, P.; Rosario, A.M.; Golde, T.E.; Li, Q. Amyloid beta peptides overexpression in retinal pigment epithelial cells via AAV-mediated gene transfer mimics AMD-like pathology in mice. Sci. Rep. 2017, 7, 3222.
- Rong, S.S.; Lee, B.Y.; Kuk, A.K.; Yu, X.T.; Li, S.S.; Li, J.; Guo, Y.; Yin, Y.; Osterbur, D.L.; Yam, J.C.S.; et al. Comorbidity of dementia and age-related macular degeneration calls for clinical awareness: A meta-analysis. Br. J. Ophthalmol. 2019, 103, 1777–1783.
- Striebel, J.F.; Race, B.; Leung, J.M.; Schwartz, C.; Chesebro, B. Prion-induced photoreceptor degeneration begins with misfolded prion protein accumulation in cones at two distinct sites: Cilia and ribbon synapses. Acta Neuropathol. Commun. 2021, 9, 17.
- Ornek, N.; Dag, E.; Ornek, K. Corneal sensitivity and tear function in neurodegenerative diseases. Curr. Eye Res. 2015, 40, 423–428.
- Dong, Z.; Luo, A.; Gan, Y.; Li, J. Amyloid Beta Deposition Could Cause Corneal Epithelial Cell Degeneration Associated with Increasing Apoptosis in APPswePS1 Transgenic Mice. Curr. Eye Res. 2018, 43, 1326–1333.
- Lee, E.J.; Kim, T.W.; Lee, D.S.; Kim, H.; Park, Y.H.; Kim, J.; Lee, J.W.; Kim, S. Increased CSF tau level is correlated with decreased lamina cribrosa thickness. Alzheimers Res. Ther. 2016, 8, 6.
- Dragan, M.C.; Leonard, T.K.; Lozano, A.M.; McAndrews, M.P.; Ng, K.; Ryan, J.D.; Tang-Wai, D.F.; Wynn, J.S.; Hoffman, K.L. Pupillary responses and memory-guided visual search reveal age-related and Alzheimer’s-related memory decline. Behav. Brain Res. 2017, 322, 351–361.
- Iijima, A.; Haida, M.; Ishikawa, N.; Ueno, A.; Minamitani, H.; Shinohara, Y. Re-evaluation of tropicamide in the pupillary response test for Alzheimer’s disease. Neurobiol. Aging 2003, 24, 789–796.
- Scinto, L.F. ApoE allelic variability influences pupil response to cholinergic challenge and cognitive impairment. Genes. Brain Behav. 2007, 6, 209–215.
- Kawasaki, A.; Ouanes, S.; Crippa, S.V.; Popp, J. Early-Stage Alzheimer’s Disease Does Not Alter Pupil Responses to Colored Light Stimuli. J. Alzheimers Dis. 2020, 75, 1273–1282.
- Kremen, W.S.; Panizzon, M.S.; Elman, J.A.; Granholm, E.L.; Andreassen, O.A.; Dale, A.M.; Gillespie, N.A.; Gustavson, D.E.; Logue, M.W.; Lyons, M.J.; et al. Pupillary dilation responses as a midlife indicator of risk for Alzheimer’s disease: Association with Alzheimer’s disease polygenic risk. Neurobiol. Aging 2019, 83, 114–121.
- Kerbage, C.; Sadowsky, C.H.; Jennings, D.; Cagle, G.D.; Hartung, P.D. Alzheimer’s disease diagnosis by detecting exogenous fluorescent signal of ligand bound to Beta amyloid in the lens of human eye: An exploratory study. Front. Neurol. 2013, 4, 62.
- Melov, S.; Wolf, N.; Strozyk, D.; Doctrow, S.R.; Bush, A.I. Mice transgenic for Alzheimer disease beta-amyloid develop lens cataracts that are rescued by antioxidant treatment. Free Radic. Biol. Med. 2005, 38, 258–261.
- Sun, S.W.; Nishioka, C.; Labib, W.; Liang, H.F. Axonal Terminals Exposed to Amyloid-beta May Not Lead to Pre-Synaptic Axonal Damage. J. Alzheimers Dis. 2015, 45, 1139–1148.
- Nishioka, C.; Liang, H.F.; Barsamian, B.; Sun, S.W. Amyloid-beta induced retrograde axonal degeneration in a mouse tauopathy model. Neuroimage 2019, 189, 180–191.
- Sadun, A.A.; Bassi, C.J. Optic nerve damage in Alzheimer’s disease. Ophthalmology 1990, 97, 9–17.
- Bayer, A.U.; Keller, O.N.; Ferrari, F.; Maag, K.P. Association of glaucoma with neurodegenerative diseases with apoptotic cell death: Alzheimer’s disease and Parkinson’s disease. Am. J. Ophthalmol. 2002, 133, 135–137.
- Wostyn, P.; Audenaert, K.; De Deyn, P.P. Alzheimer’s disease-related changes in diseases characterized by elevation of intracranial or intraocular pressure. Clin. Neurol. Neurosurg. 2008, 110, 101–109.
- Lenoir, H.; Sieroff, E. Visual perceptual disorders in Alzheimer’s disease. Geriatr. Psychol. Neuropsychiatr. Vieil. 2019, 17, 307–316.
- Wright, L.M.; Stein, T.D.; Jun, G.; Chung, J.; McConnell, K.; Fiorello, M.; Siegel, N.; Ness, S.; Xia, W.; Turner, K.L.; et al. Association of Cognitive Function with Amyloid-beta and Tau Proteins in the Vitreous Humor. J. Alzheimers Dis. 2019, 68, 1429–1438.
- Hart, N.J.; Koronyo, Y.; Black, K.L.; Koronyo-Hamaoui, M. Ocular indicators of Alzheimer’s: Exploring disease in the retina. Acta Neuropathol. 2016, 132, 767–787.
- Ho, W.L.; Leung, Y.; Tsang, A.W.; So, K.F.; Chiu, K.; Chang, R.C. Review: Tauopathy in the retina and optic nerve: Does it shadow pathological changes in the brain? Mol. Vis. 2012, 18, 2700–2710.
- Chiasseu, M.; Cueva Vargas, J.L.; Destroismaisons, L.; Vande Velde, C.; Leclerc, N.; Di Polo, A. Tau Accumulation, Altered Phosphorylation, and Missorting Promote Neurodegeneration in Glaucoma. J. Neurosci. 2016, 36, 5785–5798.
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Drose, S.; Brandt, U.; et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062.
- Michael, R.; Otto, C.; Lenferink, A.; Gelpi, E.; Montenegro, G.A.; Rosandic, J.; Tresserra, F.; Barraquer, R.I.; Vrensen, G.F. Absence of amyloid-beta in lenses of Alzheimer patients: A confocal Raman microspectroscopic study. Exp. Eye Res. 2014, 119, 44–53.
- Choi, S.I.; Lee, B.; Woo, J.H.; Jeong, J.B.; Jun, I.; Kim, E.K. APP processing and metabolism in corneal fibroblasts and epithelium as a potential biomarker for Alzheimer’s disease. Exp. Eye Res. 2019, 182, 167–174.
- Einarsdottir, A.B.; Hardarson, S.H.; Kristjansdottir, J.V.; Bragason, D.T.; Snaedal, J.; Stefansson, E. Retinal oximetry imaging in Alzheimer’s disease. J. Alzheimers Dis. 2016, 49, 79–83.
- Olafsdottir, O.B.; Saevarsdottir, H.S.; Hardarson, S.H.; Hannesdottir, K.H.; Traustadottir, V.D.; Karlsson, R.A.; Einarsdottir, A.B.; Jonsdottir, K.D.; Stefansson, E.; Snaedal, J. Retinal oxygen metabolism in patients with mild cognitive impairment. Alzheimers Dement 2018, 10, 340–345.
- Koronyo, Y.; Salumbides, B.C.; Black, K.L.; Koronyo-Hamaoui, M. Alzheimer’s disease in the retina: Imaging retinal abeta plaques for early diagnosis and therapy assessment. Neurodegener. Dis. 2012, 10, 285–293.
- Cheung, C.Y.; Ong, Y.T.; Ikram, M.K.; Ong, S.Y.; Li, X.; Hilal, S.; Catindig, J.A.; Venketasubramanian, N.; Yap, P.; Seow, D.; et al. Microvascular network alterations in the retina of patients with Alzheimer’s disease. Alzheimers Dement. 2014, 10, 135–142.
- Frost, S.; Kanagasingam, Y.; Sohrabi, H.; Vignarajan, J.; Bourgeat, P.; Salvado, O.; Villemagne, V.; Rowe, C.C.; Macaulay, S.L.; Szoeke, C.; et al. Retinal vascular biomarkers for early detection and monitoring of Alzheimer’s disease. Transl. Psychiatry 2013, 3, e233.
- Shi, H.; Koronyo, Y.; Rentsendorj, A.; Regis, G.C.; Sheyn, J.; Fuchs, D.T.; Kramerov, A.A.; Ljubimov, A.V.; Dumitrascu, O.M.; Rodriguez, A.R.; et al. Identification of early pericyte loss and vascular amyloidosis in Alzheimer’s disease retina. Acta Neuropathol. 2020, 139, 813–836.
- Liu, S.S.; Zhu, S.Q. . Zhonghua Yan Ke Za Zhi 2017, 53, 314–316.
- Frederikse, P.H.; Zigler, J.S., Jr. Presenilin expression in the ocular lens. Curr. Eye Res. 1998, 17, 947–952.
- Ho, C.Y.; Troncoso, J.C.; Knox, D.; Stark, W.; Eberhart, C.G. Beta-amyloid, phospho-tau and alpha-synuclein deposits similar to those in the brain are not identified in the eyes of Alzheimer’s and Parkinson’s disease patients. Brain Pathol. 2014, 24, 25–32.
- Kwak, D.E.; Ko, T.; Koh, H.S.; Ji, Y.W.; Shin, J.; Kim, K.; Kim, H.Y.; Lee, H.K.; Kim, Y. Alterations of aqueous humor Abeta levels in Abeta-infused and transgenic mouse models of Alzheimer disease. PLoS ONE 2020, 15, e0227618.
- Prakasam, A.; Muthuswamy, A.; Ablonczy, Z.; Greig, N.H.; Fauq, A.; Rao, K.J.; Pappolla, M.A.; Sambamurti, K. Differential accumulation of secreted AbetaPP metabolites in ocular fluids. J. Alzheimers Dis. 2010, 20, 1243–1253.
- Cunha, J.P.; Proenca, R.; Dias-Santos, A.; Melancia, D.; Almeida, R.; Aguas, H.; Santos, B.O.; Alves, M.; Ferreira, J.; Papoila, A.L.; et al. Choroidal thinning: Alzheimer’s disease and aging. Alzheimers Dement 2017, 8, 11–17.
- Trebbastoni, A.; Marcelli, M.; Mallone, F.; D’Antonio, F.; Imbriano, L.; Campanelli, A.; de Lena, C.; Gharbiya, M. Attenuation of Choroidal Thickness in Patients with Alzheimer Disease: Evidence From an Italian Prospective Study. Alzheimer Dis. Assoc. Disord. 2017, 31, 128–134.
- Rizzo, M.; Anderson, S.W.; Dawson, J.; Nawrot, M. Vision and cognition in Alzheimer’s disease. Neuropsychologia 2000, 38, 1157–1169.
- Chiu, K.; Chan, T.F.; Wu, A.; Leung, I.Y.; So, K.F.; Chang, R.C. Neurodegeneration of the retina in mouse models of Alzheimer’s disease: What can we learn from the retina? Age 2012, 34, 633–649.
- Guo, L.; Salt, T.E.; Luong, V.; Wood, N.; Cheung, W.; Maass, A.; Ferrari, G.; Russo-Marie, F.; Sillito, A.M.; Cheetham, M.E.; et al. Targeting amyloid-beta in glaucoma treatment. Proc. Natl. Acad. Sci. USA 2007, 104, 13444–13449.
- Edwards, M.M.; Rodriguez, J.J.; Gutierrez-Lanza, R.; Yates, J.; Verkhratsky, A.; Lutty, G.A. Retinal macroglia changes in a triple transgenic mouse model of Alzheimer’s disease. Exp. Eye Res. 2014, 127, 252–260.
- Joly, S.; Lamoureux, S.; Pernet, V. Nonamyloidogenic processing of amyloid beta precursor protein is associated with retinal function improvement in aging male APP(swe)/PS1DeltaE9 mice. Neurobiol. Aging 2017, 53, 181–191.
- Walsh, D.T.; Bresciani, L.; Saunders, D.; Manca, M.F.; Jen, A.; Gentleman, S.M.; Jen, L.S. Amyloid beta peptide causes chronic glial cell activation and neuro-degeneration after intravitreal injection. Neuropathol. Appl. Neurobiol. 2005, 31, 491–502.
- Chiasseu, M.; Alarcon-Martinez, L.; Belforte, N.; Quintero, H.; Dotigny, F.; Destroismaisons, L.; Vande Velde, C.; Panayi, F.; Louis, C.; Di Polo, A. Tau accumulation in the retina promotes early neuronal dysfunction and precedes brain pathology in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 58.
- Jarosz-Griffiths, H.H.; Noble, E.; Rushworth, J.V.; Hooper, N.M. Amyloid-beta Receptors: The Good, the Bad, and the Prion Protein. J. Biol. Chem. 2016, 291, 3174–3183.
- Lauren, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132.
- Nygaard, H.B.; van Dyck, C.H.; Strittmatter, S.M. Fyn kinase inhibition as a novel therapy for Alzheimer’s disease. Alzheimers Res. Ther. 2014, 6, 8.
- Altmeppen, H.C.; Puig, B.; Dohler, F.; Thurm, D.K.; Falker, C.; Krasemann, S.; Glatzel, M. Proteolytic processing of the prion protein in health and disease. Am. J. Neurodegener. Dis. 2012, 1, 15–31.
- Asai, M.; Hattori, C.; Szabo, B.; Sasagawa, N.; Maruyama, K.; Tanuma, S.; Ishiura, S. Putative function of ADAM9, ADAM10, and ADAM17 as APP alpha-secretase. Biochem. Biophys. Res. Commun. 2003, 301, 231–235.
- Asthana, A.; Baksi, S.; Ashok, A.; Karmakar, S.; Mammadova, N.; Kokemuller, R.; Greenlee, M.H.; Kong, Q.; Singh, N. Prion protein facilitates retinal iron uptake and is cleaved at the beta-site: Implications for retinal iron homeostasis in prion disorders. Sci. Rep. 2017, 7, 9600.
- Castle, A.R.; Gill, A.C. Physiological Functions of the Cellular Prion Protein. Front. Mol. Biosci. 2017, 4, 19.
- Ezpeleta, J.; Boudet-Devaud, F.; Pietri, M.; Baudry, A.; Baudouin, V.; Alleaume-Butaux, A.; Dagoneau, N.; Kellermann, O.; Launay, J.M.; Schneider, B. Protective role of cellular prion protein against TNFalpha-mediated inflammation through TACE alpha-secretase. Sci. Rep. 2017, 7, 7671.
- Singh, N. The role of iron in prion disease and other neurodegenerative diseases. PLoS Pathog. 2014, 10, e1004335.
- Singh, N.; Singh, A.; Das, D.; Mohan, M.L. Redox control of prion and disease pathogenesis. Antioxid. Redox Signal 2010, 12, 1271–1294.
- Ramana, K.V. Tumor necrosis factor-alpha converting enzyme: Implications for ocular inflammatory diseases. Int. J. Biochem. Cell Biol. 2010, 42, 1076–1079.
- Frigg, R.; Wenzel, A.; Samardzija, M.; Oesch, B.; Wariwoda, H.; Navarini, A.A.; Seeliger, M.W.; Tanimoto, N.; Reme, C.; Grimm, C. The prion protein is neuroprotective against retinal degeneration in vivo. Exp. Eye Res. 2006, 83, 1350–1358.
- Loubet, D.; Dakowski, C.; Pietri, M.; Pradines, E.; Bernard, S.; Callebert, J.; Ardila-Osorio, H.; Mouillet-Richard, S.; Launay, J.M.; Kellermann, O.; et al. Neuritogenesis: The prion protein controls beta1 integrin signaling activity. FASEB J. 2012, 26, 678–690.
- Alleaume-Butaux, A.; Nicot, S.; Pietri, M.; Baudry, A.; Dakowski, C.; Tixador, P.; Ardila-Osorio, H.; Haeberle, A.M.; Bailly, Y.; Peyrin, J.M.; et al. Double-Edge Sword of Sustained ROCK Activation in Prion Diseases through Neuritogenesis Defects and Prion Accumulation. PLoS Pathog. 2015, 11, e1005073.
- Kim, H.J.; Choi, H.S.; Park, J.H.; Kim, M.J.; Lee, H.G.; Petersen, R.B.; Kim, Y.S.; Park, J.B.; Choi, E.K. Regulation of RhoA activity by the cellular prion protein. Cell Death Dis. 2017, 8, e2668.
- Bertuchi, F.R.; Bourgeon, D.M.; Landemberger, M.C.; Martins, V.R.; Cerchiaro, G. PrPC displays an essential protective role from oxidative stress in an astrocyte cell line derived from PrPC knockout mice. Biochem. Biophys. Res. Commun. 2012, 418, 27–32.
- Dupiereux, I.; Falisse-Poirrier, N.; Zorzi, W.; Watt, N.T.; Thellin, O.; Zorzi, D.; Pierard, O.; Hooper, N.M.; Heinen, E.; Elmoualij, B. Protective effect of prion protein via the N-terminal region in mediating a protective effect on paraquat-induced oxidative injury in neuronal cells. J. Neurosci. Res. 2008, 86, 653–659.
- Haigh, C.L.; Collins, S.J. Endoproteolytic cleavage as a molecular switch regulating and diversifying prion protein function. Neural Regen. Res. 2016, 11, 238–239.
- Zeng, F.; Watt, N.T.; Walmsley, A.R.; Hooper, N.M. Tethering the N-terminus of the prion protein compromises the cellular response to oxidative stress. J. Neurochem. 2003, 84, 480–490.
- Zeng, L.; Zou, W.; Wang, G. Cellular prion protein (PrP(C)) and its role in stress responses. Int. J. Clin. Exp. Med. 2015, 8, 8042–8050.
- Brown, D.R.; Clive, C.; Haswell, S.J. Antioxidant activity related to copper binding of native prion protein. J. Neurochem. 2001, 76, 69–76.
- Singh, A.; Haldar, S.; Horback, K.; Tom, C.; Zhou, L.; Meyerson, H.; Singh, N. Prion protein regulates iron transport by functioning as a ferrireductase. J. Alzheimers Dis. 2013, 35, 541–552.
- Wille, H.; Requena, J.R. The Structure of PrP(Sc) Prions. Pathogens 2018, 7, 20.
- Ramirez, A.I.; de Hoz, R.; Salobrar-Garcia, E.; Salazar, J.J.; Rojas, B.; Ajoy, D.; Lopez-Cuenca, I.; Rojas, P.; Trivino, A.; Ramirez, J.M. The Role of Microglia in Retinal Neurodegeneration: Alzheimer’s Disease, Parkinson, and Glaucoma. Front. Aging Neurosci. 2017, 9, 214.
- Surguchov, A.; McMahan, B.; Masliah, E.; Surgucheva, I. Synucleins in ocular tissues. J. Neurosci. Res. 2001, 65, 68–77.