Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Peter Tang and Version 1 by Thierry Coppola.
Among the most widespread pathologies, obesity, which is often associated with diabetes, is constantly increasing in incidence, and in parallel, neurodegenerative and mood disorders are increasingly affecting many people. For years, these pathologies have been so frequently observed in the population in a concomitant way that they are considered as comorbidities. In fact, common mechanisms are certainly at work in the etiology of these pathologies. Fluoxetine was discovered in the 1970s. Initially called LY110140, it was described as a selective 5-HT reuptake inhibitor.
- depression
- diabetes
- fluoxetine
1. Introduction
It is generally accepted that a drug’s pleiotropy is observed when its effects are different from those initially intended. This includes both negative and positive effects. Up to now, research into and the development of treatments for a given pathology have generated drugs which, when used, have revealed multiple side effects, some of them deleterious, leading to more restrictive use or abandonment. There are numerous examples, particularly for chronic treatments, such as those employed in diabetes and depression [1,2][1][2]. In many cases, the collateral clinical effects are mediated by mechanisms other than those associated with the target for which the drug was initially designed. Collateral clinical effects are often justified by the presence of the initially identified target in another organ, tissue or cell. Both scenarios raise concerns about pleiotropic effects and emphasize the necessity for a comprehensive characterization of the various molecular impacts of the drug, especially those linked to its target. Rather than considering pleiotropy as a more or less beneficial inevitability, wthe researchers are seeking to highlight the value of using one of its aspects, i.e., targeting a molecular target present in different tissues, to seek convergence toward specifically beneficial effects. Here, wethe researchers seek to illustrate this point of view by taking stock of knowledge on two pathologies that are often linked: diabetes and depression.
Depression is the most common psychiatric pathology, with a prevalence that is estimated to range from 5% to 20% of the general population [3]. Depressive disorders are characterized by sadness of sufficient severity or persistence that interferes with daily living and often by diminished interest or pleasure in activities (anhedonia). The exact cause is unknown but is probably multifactorial, involving heredity, altered neurotransmitter levels, altered neuroendocrine functions and psychosocial factors. Therapeutic approach usually involves medication and/or psychotherapy. Depressive states are often associated with deficits in serotonin (5-hydroxytryptamine, 5-HT), which is an essential neurotransmitter for communication between neurons and is involved in eating, sexual behavior, the sleep–wake cycle, pain and anxiety or mood disorders [4,5][4][5]. Being defined by the World Health Organization as a common mental disorder worldwide, depression is the main mental disability leading to death (WHO, 2021) [6]. Numerous reports suggest that 2/3 of individuals taking antidepressant drugs actually benefit from their medications. However, for the remaining 1/3, antidepressants currently available on the market are ineffective and/or make their depressive symptoms worse [7].
Diet-Induced Obesity (DIO) and type 2 diabetes mellitus (T2DM) also represent major healthcare problems. DIO alone was identified as the cause of 80% of all T2DM cases, and both disorders mainly result from adverse eating habits and inadequate physical activity. Although there is an abundance of research examining the complex association between obesity and major depressive disorder (MDD), the conclusions are still inconsistent [8]. Whereas the larger body of evidence is leaning toward the presence of a link between these two pathological conditions [9], several studies report that they are unrelated [10] or only show an association in subgroups, for example, in women [11]. A review [9] summarizing the epidemiological evidence of the interconnection between obesity and MDD from large meta-analyses suggests overall that obesity and depression are bi-directionally associated, with the presence of one increasing the risk of developing the other. Overall, the rate of mild, moderate and severe depression in patients with diabetes increases with a higher body mass index (BMI). Subjects with obesity and diabetes appear to be at an even higher risk for depression compared to subjects with obesity but not diabetes [12]. Thus, subjects with obesity and diabetes are at greater risk of depression compared to the general population.
2. Depression and Diabetes as Comorbidities
As mentioned above, obesity can increase the risk of depression, and depression is predictive of developing obesity. Psychological stresses frequently lead to modifications of hormone levels and proinflammatory molecules (C-reactive protein and cytokines) that generate a higher risk of type 2 diabetes and depression [16][13]. Both overall adiposity (total body fat and BMI) and abdominal adiposity (waist circumference and visceral adipose) measures are associated with depressive mood. The strongest association is observed between levels of adiposity and specific “atypical” neurovegetative depressive mood symptoms (e.g., fatigability and hyperphagia), which may be an indication of an alteration in the energy homeostasis. A higher degree of obesity is likely causal for the specific symptom of increased appetite in participants with depression. Indeed, subjects with atypical depression have markedly elevated obesity rates compared to population controls and to other subjects with depression [17][14]. In contrast, obesity rates are not significantly different in subjects with classic depression and controls without depression. Thus, refining the target phenotype(s) for future work on depression and obesity might improve our understanding, prevention and treatment of this complex clinical problem [18][15]. There is also several established molecular links between depressive pathology and some adipose-related metabolic signals such as glucocorticoids, leptin, adiponectin, resistin, insulin and inflammatory signals [16][13]. Elevated glucocorticoids levels, produced by adrenal glands, are implicated in the pathophysiology of both obesity and depression. Indeed, the critical role of corticoids on adipose tissue deposition was demonstrated by studies showing that adrenalectomy prevents obesity [19][16]. In addition, repeated administration of corticosterone to rodents is reported to display depressive-like behavior [20][17], and leptin-deficient ob/ob mice (obese and hyperglycemic animals) have elevated corticosterone, which is reduced by leptin treatment [21][18]. Genetic analysis of risk factors for MDD and T2DM almost expectedly shows an association of comorbidity genes involved in natural immunity or cellular aging [22][19]. Genes relevant to the innate immune system, tau protein formation and cellular aging were identified, and the experimental results indicate that the common, often comorbid, conditions of MDD and T2DM have a common molecular pathway [22][19]. For example, overexpression of the BDNF gene in the dorsal raphe nucleus (DRN) of obese and diabetic mice subjected to a stress-induced depression protocol will have an associated antidepressant effect by improving serotonin homeostasis [23][20]. In addition, the improvement in metabolic biological constants due to BDNF overexpression shows, as expected, the importance of the DRN in depression as well as the importance of this brain area in diabetes [23][20]. Nowadays, various therapeutic approaches are proposed for patients with depression and diabetes. Among these, drugs targeting a specific receptor or channel at the transmembrane level have proven effectiveness, bringing them to the level of major therapeutic approaches such as fluoxetine for treating depression and GLP-1 as an antidiabetic. Moreover, besides their original expected effect, their clinical long-lasting use has revealed unexpected additional beneficial effects.3. Fluoxetine
3.1. Fluoxetine as an Antidepressant Reference Treatment
Since the 1960s, the strategies based on antidepressant molecule development have mainly focused on increasing the quantity of 5-HT released in the synaptic cleft, the space between two neurons where nerve communications take place via neurotransmitters. 5-HT can activate the different subtypes of the 5-HT receptor family (1, 2, 3, 4, 5, 6 and 7), leading to their respective signal transduction pathway within the postsynaptic neurons [24][21]. In presynaptic 5-HT terminals, 5-HT is either taken up by storage vesicles through selective serotonin transporters or degraded by monoamine oxidase (MAO). Some MDD treatments target the serotoninergic system through two pharmacological approaches: selective 5-HT1A receptor antagonists [25][22] and selective serotonin reuptake inhibitors (SSRIs), including fluoxetine [26][23]. This latter class of antidepressants is considered serotoninergic because they increase intrasynaptic serotonin concentrations by inhibiting presynaptic 5-HT reuptake, leading to the stimulation of postsynaptic 5-HT receptors. Thus, the serotonin remaining at the synaptic cleft for a longer period of time would repeatedly stimulate the receptors of the postsynaptic cell. Fluoxetine was discovered in the 1970s. Initially called LY110140, it was described as a selective 5-HT reuptake inhibitor [27,28][24][25]. Fluoxetine hydrochloride (better known as ProzacR) was the first molecule in the family of antidepressants known as SSRIs [29][26], the most widely prescribed antidepressants for the treatment of depressive states nowadays. The first clinical study conducted in 1993 showed its efficacy on severe depression with few side effects, allowing its use for long-term treatment [30][27]. It took several years to demonstrate the physical interaction between the serotonin transporter (SERT) and fluoxetine. Indeed, at the molecular level, SSRIs bind directly to the SERT to maintain the transporter in an outward open conformation, preventing the binding of substrates [31][28]. SSRIs are selective for the serotonergic 5-HT system but not specific for a particular 5-HT receptor. Indeed, they allow stimulation of 5-HT1 receptors, combining antidepressant and anxiolytic effects, as well as that of 5-HT2, often causing anxiety, insomnia and sexual dysfunction, and 5-HT3 receptors, inducing nausea and headache. Thus, selective serotonin reuptake inhibitors can paradoxically relieve and generate anxiety. In addition, it was shown very recently that antidepressant drugs were binding directly to the TRKB neurotrophin receptor, which facilitates BDNF stimulation [32][29]. This new piece of knowledge highlights the complex effects of SSRI drugs and strengthens the evidence for their potential pleiotropic action.3.2. Fluoxetine Action on Pancreatic Endocrine Function
In the clinic, it became apparent early on that fluoxetine could be used on very large patient populations. Patients with diabetes are as sensitive to fluoxetine as patients with depression, and this treatment also improves glycemia after only few weeks [33,34,35][30][31][32]. Indeed, fluoxetine tends to improve glycemic regulation and weight loss by inducing higher insulin sensitivity and regulation of skeletal muscle glycogen synthase activity [36][33]. In fact, this increase in insulin sensitivity seems to be one of the major effects observed among patients treated with Prozac [37][34]. Fluoxetine significantly reduces food intake in lean or obese rats [38[35][36],39], and its indirect effect on weight maintenance is achieved by the balance between food intake and energy expenditure managed by the hypothalamus [40][37]. Interestingly, an atlas of vagal sensory neurons has recently been published. The authors indicate that serotonin is expressed in specific neuron types [41][38]. This suggests a possible peripheral effect of fluoxetine on these neurons, which innervate the pancreas. In this vein, in mice, electrostimulation of the pancreatic nerve has been shown to be an effective approach to eradicating recent-onset type 1 diabetes [42][39]. Interestingly, serotonin is expressed in endocrine cells of the pancreas and is sequestered in the same secretory granules as pancreatic hormones [43,44][40][41]. Whereas on one hand 5HT regulates the pancreatic secretion of glucagon and insulin (32), 5-HT secretion is, on another hand, regulated by glucose in β-cell lines (MIN6) in vitro [45][42] and vesicular transporters of 5-HT (VMAT1/2) are expressed in pancreatic β cells [46][43]. In addition, the increase in β-cell mass during gestation requires the control of serotonin homeostasis. Thus, altered serotonin signaling also contributes to β-cell mass dysfunction and to diabetes [43][40]. The clearance transporter SERT is also expressed in β cells [47][44], suggesting an effect of fluoxetine on the endocrine pancreas. Recently, a direct effect of fluoxetine on pancreatic β cells to potentiate insulin exocytosis has been shown [48][45], and preliminary experiments on ob/ob mice show improvement in metabolic physiological parameters [48][45]. However, several other in vitro studies have shown the opposite results [49,50,51][46][47][48]. For example, insulin secretion is inhibited by fluoxetine [49,51][46][48] in rodent and [47][44] in human islets. Additionally, the increase in serotonin concentration outside the β cells induces the dysfunction of mitochondrial activity, which is by itself coupled to insulin secretion [49][46]. Thus, despite the extensive research conducted in the field, the effects of 5-HT on the endocrine pancreas remain difficult to grasp because of the expression of various receptors and transporters of 5-HT in the islets of Langerhans [52][49] (Figure 1). For now, the molecular data allow us to anticipate a long-term effect of treatments using SSRIs, as shown in animal model studies [53][50]. The complex effects of serotonin on the adaptive mechanisms of the endocrine pancreas suggest that caution is required in the use of drugs targeting this signaling system.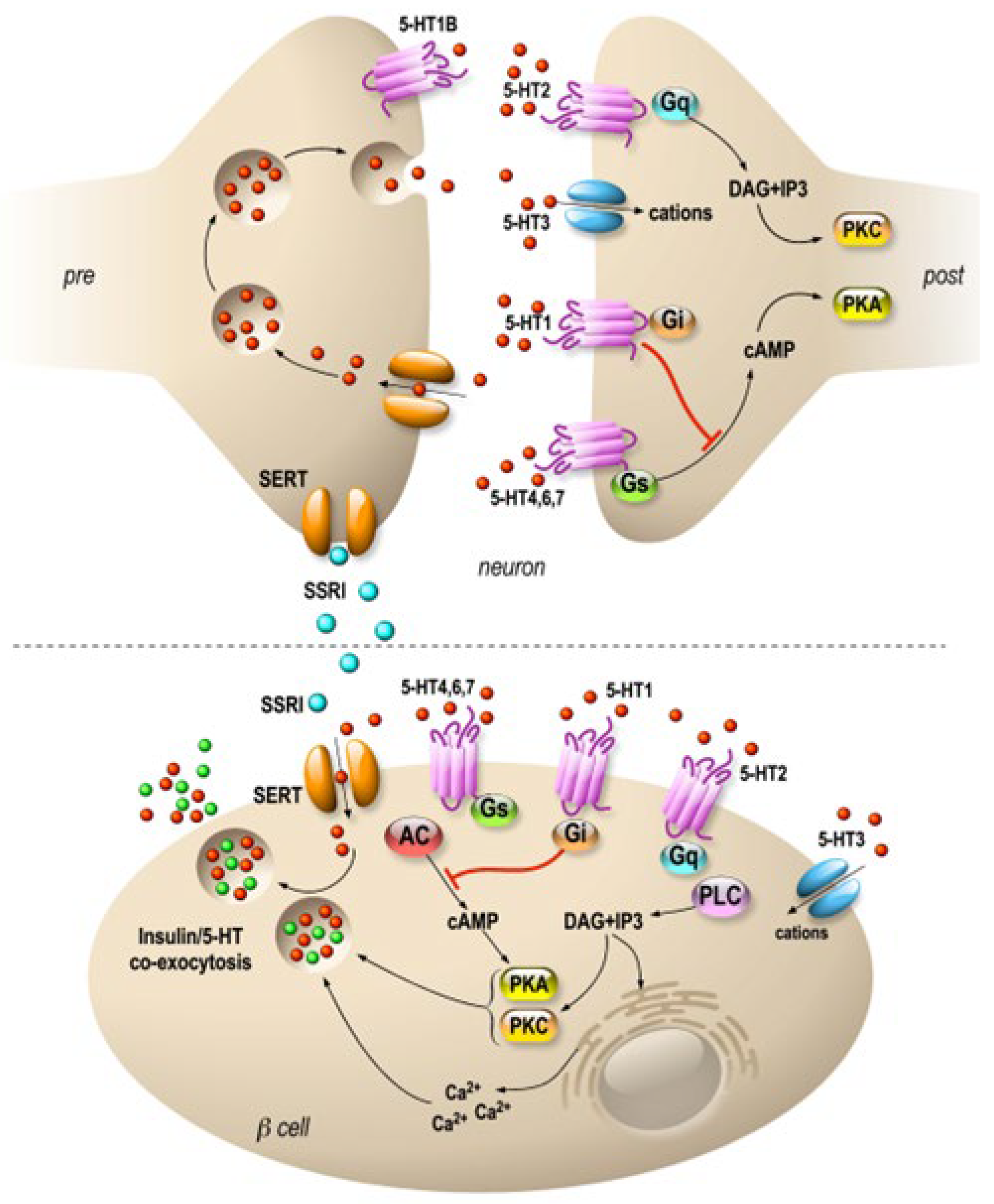
Figure 1. Specific molecular targets of fluoxetine on neurons and pancreatic β cells. Distribution of 5-HT transporters and receptors in neurons (upper panel) and in pancreatic β cells (lower panel). In neurons, 5-HT receptors are expressed mostly at the postsynaptic level, and they modulate signal transmission. β cells express 5-HT receptors similar to those of neurons. SSRIs are symbolized by blue, 5-HT by red and insulin by green dots. 5-HT1, 2, 4, 6 and 7 are 7-TM domain receptors (GPCRs). 5-HT3 is a cationic channel symbolized by blue. SERTs are in orange.
References
- Brady, P.A.; Terzic, A. The sulfonylurea controversy: More questions from the heart. J. Am. Coll. Cardiol. 1998, 31, 950–956.
- Goodnick, P.J.; Henry, J.H.; Buki, V.M. Treatment of depression in patients with diabetes mellitus. J. Clin. Psychiatry 1995, 56, 128–136.
- Herrman, H.; Patel, V.; Kieling, C.; Berk, M.; Buchweitz, C.; Cuijpers, P.; Furukawa, T.A.; Kessler, R.C.; Kohrt, B.A.; Maj, M.; et al. Time for united action on depression: A Lancet-World Psychiatric Association Commission. Lancet 2022, 399, 957–1022.
- Coppen, A. The biochemistry of affective disorders. Br. J. Psychiatry 1967, 113, 1237–1264.
- Delgado, P.L. Depression: The case for a monoamine deficiency. J. Clin. Psychiatry 2000, 61 (Suppl. S6), 7–11.
- Plana-Ripoll, O.; Pedersen, C.B.; Agerbo, E.; Holtz, Y.; Erlangsen, A.; Canudas-Romo, V.; Andersen, P.K.; Charlson, F.J.; Christensen, M.K.; Erskine, H.E.; et al. A comprehensive analysis of mortality-related health metrics associated with mental disorders: A nationwide, register-based cohort study. Lancet 2019, 394, 1827–1835.
- Miola, A.; Meda, N.; Perini, G.; Sambataro, F. Structural and functional features of treatment-resistant depression: A systematic review and exploratory coordinate-based meta-analysis of neuroimaging studies. Psychiatry Clin. Neurosci. 2023, 77, 252–263.
- de Wit, L.M.; Fokkema, M.; van Straten, A.; Lamers, F.; Cuijpers, P.; Penninx, B.W. Depressive and anxiety disorders and the association with obesity, physical, and social activities. Depress. Anxiety 2010, 27, 1057–1065.
- Milaneschi, Y.; Simmons, W.K.; van Rossum, E.F.C.; Penninx, B.W. Depression and obesity: Evidence of shared biological mechanisms. Mol. Psychiatry 2019, 24, 18–33.
- Delahanty, L.M.; Meigs, J.B.; Hayden, D.; Williamson, D.A.; Nathan, D.M.; Diabetes Prevenion Program Research, G. Psychological and behavioral correlates of baseline BMI in the diabetes prevention program (DPP). Diabetes Care 2002, 25, 1992–1998.
- Heo, M.; Pietrobelli, A.; Fontaine, K.R.; Sirey, J.A.; Faith, M.S. Depressive mood and obesity in US adults: Comparison and moderation by sex, age, and race. Int. J. Obes. 2006, 30, 513–519.
- Lee, J.; Kim, K.H.; Ahn, J.C.; Kim, J.A.; Lee, G.; Son, J.S.; Choi, S.J.; Oh, Y.H.; Park, S.M. Prevalence, awareness, treatment, and control of diabetes mellitus by depressive symptom severity: A cross-sectional analysis of NHANES 2011-2016. BMJ Open Diabetes Res. Care 2021, 9, e002268.
- Hryhorczuk, C.; Sharma, S.; Fulton, S.E. Metabolic disturbances connecting obesity and depression. Front. Neurosci. 2013, 7, 177.
- Alshehri, T.; Boone, S.; de Mutsert, R.; Penninx, B.; Rosendaal, F.; le Cessie, S.; Milaneschi, Y.; Mook-Kanamori, D. The association between overall and abdominal adiposity and depressive mood: A cross-sectional analysis in 6459 participants. Psychoneuroendocrinology 2019, 110, 104429.
- Levitan, R.D.; Davis, C.; Kaplan, A.S.; Arenovich, T.; Phillips, D.I.; Ravindran, A.V. Obesity comorbidity in unipolar major depressive disorder: Refining the core phenotype. J. Clin. Psychiatry 2012, 73, 1119–1124.
- Freedman, M.R.; Castonguay, T.W.; Stern, J.S. Effect of adrenalectomy and corticosterone replacement on meal patterns of Zucker rats. Am. J. Physiol. 1985, 249, R584–R594.
- Gregus, A.; Wintink, A.J.; Davis, A.C.; Kalynchuk, L.E. Effect of repeated corticosterone injections and restraint stress on anxiety and depression-like behavior in male rats. Behav. Brain Res. 2005, 156, 105–114.
- Liu, Y.; Nakagawa, Y.; Wang, Y.; Li, R.; Li, X.; Ohzeki, T.; Friedman, T.C. Leptin activation of corticosterone production in hepatocytes may contribute to the reversal of obesity and hyperglycemia in leptin-deficient ob/ob mice. Diabetes 2003, 52, 1409–1416.
- Liu, D.; McIntyre, R.S.; Li, R.; Yang, M.; Xue, Y.; Cao, B. Genetic association between major depressive disorder and type 2 diabetes mellitus: Shared pathways and protein networks. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 111, 110339.
- Xiu, J.; Han, R.; Liu, Z.; Li, J.; Liu, S.; Shen, Y.; Ding, Y.Q.; Xu, Q. Hijacking Dorsal Raphe to Improve Metabolism and Depression-like Behaviors via BDNF Gene Transfer in Mice. Diabetes 2021, 70, 1780–1793.
- Barnes, N.M.; Ahern, G.P.; Becamel, C.; Bockaert, J.; Camilleri, M.; Chaumont-Dubel, S.; Claeysen, S.; Cunningham, K.A.; Fone, K.C.; Gershon, M.; et al. International Union of Basic and Clinical Pharmacology. CX. Classification of Receptors for 5-hydroxytryptamine; Pharmacology and Function. Pharmacol. Rev. 2021, 73, 310–520.
- Celada, P.; Bortolozzi, A.; Artigas, F. Serotonin 5-HT1A receptors as targets for agents to treat psychiatric disorders: Rationale and current status of research. CNS Drugs 2013, 27, 703–716.
- Celada, P.; Puig, M.; Amargos-Bosch, M.; Adell, A.; Artigas, F. The therapeutic role of 5-HT1A and 5-HT2A receptors in depression. J. Psychiatry Neurosci. 2004, 29, 252–265.
- Wong, D.T.; Horng, J.S.; Bymaster, F.P.; Hauser, K.L.; Molloy, B.B. A selective inhibitor of serotonin uptake: Lilly 110140, 3-(p-trifluoromethylphenoxy)-N-methyl-3-phenylpropylamine. Life Sci. 1974, 15, 471–479.
- Fuller, R.W.; Perry, K.W.; Wong, D.T.; Molloy, B.B. Effects of some homologues of 4-chloroamphetamine on brain serotonin metabolism. Neuropharmacology 1974, 13, 609–614.
- Fuller, R.W.; Wong, D.T.; Robertson, D.W. Fluoxetine, a selective inhibitor of serotonin uptake. Med. Res. Rev. 1991, 11, 17–34.
- Stokes, P.E. Fluoxetine: A five-year review. Clin. Ther. 1993, 15, 216–243, discussion 215.
- Coleman, J.A.; Green, E.M.; Gouaux, E. X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532, 334–339.
- Casarotto, P.C.; Girych, M.; Fred, S.M.; Kovaleva, V.; Moliner, R.; Enkavi, G.; Biojone, C.; Cannarozzo, C.; Sahu, M.P.; Kaurinkoski, K.; et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 2021, 184, 1299–1313.e1219.
- Daubresse, J.C.; Kolanowski, J.; Krzentowski, G.; Kutnowski, M.; Scheen, A.; Van Gaal, L. Usefulness of fluoxetine in obese non-insulin-dependent diabetics: A multicenter study. Obes. Res. 1996, 4, 391–396.
- Lustman, P.J.; Anderson, R.J.; Freedland, K.E.; de Groot, M.; Carney, R.M.; Clouse, R.E. Depression and poor glycemic control: A meta-analytic review of the literature. Diabetes Care 2000, 23, 934–942.
- Ye, Z.; Chen, L.; Yang, Z.; Li, Q.; Huang, Y.; He, M.; Zhang, S.; Zhang, Z.; Wang, X.; Zhao, W.; et al. Metabolic effects of fluoxetine in adults with type 2 diabetes mellitus: A meta-analysis of randomized placebo-controlled trials. PLoS ONE 2011, 6, e21551.
- Breum, L.; Bjerre, U.; Bak, J.F.; Jacobsen, S.; Astrup, A. Long-term effects of fluoxetine on glycemic control in obese patients with non-insulin-dependent diabetes mellitus or glucose intolerance: Influence on muscle glycogen synthase and insulin receptor kinase activity. Metabolism 1995, 44, 1570–1576.
- Maheux, P.; Ducros, F.; Bourque, J.; Garon, J.; Chiasson, J.L. Fluoxetine improves insulin sensitivity in obese patients with non-insulin-dependent diabetes mellitus independently of weight loss. Int. J. Obes. Relat. Metab. Disord. 1997, 21, 97–102.
- Dryden, S.; Frankish, H.M.; Wang, Q.; Pickavance, L.; Williams, G. The serotonergic agent fluoxetine reduces neuropeptide Y levels and neuropeptide Y secretion in the hypothalamus of lean and obese rats. Neuroscience 1996, 72, 557–566.
- Gutierrez, A.; Saracibar, G.; Casis, L.; Echevarria, E.; Rodriguez, V.M.; Macarulla, M.T.; Abecia, L.C.; Portillo, M.P. Effects of fluoxetine administration on neuropeptide y and orexins in obese zucker rat hypothalamus. Obes. Res. 2002, 10, 532–540.
- Bernardis, L.L.; Bellinger, L.L. The lateral hypothalamic area revisited: Neuroanatomy, body weight regulation, neuroendocrinology and metabolism. Neurosci. Biobehav. Rev. 1993, 17, 141–193.
- Kupari, J.; Haring, M.; Agirre, E.; Castelo-Branco, G.; Ernfors, P. An Atlas of Vagal Sensory Neurons and Their Molecular Specialization. Cell Rep. 2019, 27, 2508–2523.e2504.
- Guyot, M.; Simon, T.; Ceppo, F.; Panzolini, C.; Guyon, A.; Lavergne, J.; Murris, E.; Daoudlarian, D.; Brusini, R.; Zarif, H.; et al. Pancreatic nerve electrostimulation inhibits recent-onset autoimmune diabetes. Nat. Biotechnol. 2019, 37, 1446–1451.
- Bennet, H.; Balhuizen, A.; Medina, A.; Dekker Nitert, M.; Ottosson Laakso, E.; Essen, S.; Spegel, P.; Storm, P.; Krus, U.; Wierup, N.; et al. Altered serotonin (5-HT) 1D and 2A receptor expression may contribute to defective insulin and glucagon secretion in human type 2 diabetes. Peptides 2015, 71, 113–120.
- Gylfe, E. Association between 5-hydroxytryptamine release and insulin secretion. J. Endocrinol. 1978, 78, 239–248.
- Cataldo, L.R.; Cortes, V.A.; Mizgier, M.L.; Aranda, E.; Mezzano, D.; Olmos, P.; Galgani, J.E.; Suazo, J.; Santos, J.L. Fluoxetine impairs insulin secretion without modifying extracellular serotonin levels in MIN6 beta-cells. Exp. Clin. Endocrinol. Diabetes 2015, 123, 473–478.
- Cataldo, L.R.; Mizgier, M.L.; Busso, D.; Olmos, P.; Galgani, J.E.; Valenzuela, R.; Mezzano, D.; Aranda, E.; Cortes, V.A.; Santos, J.L. Serotonin- and Dopamine-Related Gene Expression in db/db Mice Islets and in MIN6 beta-Cells Treated with Palmitate and Oleate. J. Diabetes Res. 2016, 2016, 3793781.
- Almaca, J.; Molina, J.; Menegaz, D.; Pronin, A.N.; Tamayo, A.; Slepak, V.; Berggren, P.O.; Caicedo, A. Human Beta Cells Produce and Release Serotonin to Inhibit Glucagon Secretion from Alpha Cells. Cell Rep. 2016, 17, 3281–3291.
- Liu, B.; Ruz-Maldonado, I.; Toczyska, K.; Olaniru, O.E.; Zariwala, M.G.; Hopkins, D.; Zhao, M.; Persaud, S.J. The selective serotonin reuptake inhibitor fluoxetine has direct effects on beta cells, promoting insulin secretion and increasing beta-cell mass. Diabetes Obes. Metab. 2022, 24, 2038–2050.
- De Long, N.E.; Hyslop, J.R.; Raha, S.; Hardy, D.B.; Holloway, A.C. Fluoxetine-induced pancreatic beta cell dysfunction: New insight into the benefits of folic acid in the treatment of depression. J. Affect. Disord. 2014, 166, 6–13.
- Chang, H.Y.; Chen, S.L.; Shen, M.R.; Kung, M.L.; Chuang, L.M.; Chen, Y.W. Selective serotonin reuptake inhibitor, fluoxetine, impairs E-cadherin-mediated cell adhesion and alters calcium homeostasis in pancreatic beta cells. Sci. Rep. 2017, 7, 3515.
- Isaac, R.; Boura-Halfon, S.; Gurevitch, D.; Shainskaya, A.; Levkovitz, Y.; Zick, Y. Selective serotonin reuptake inhibitors (SSRIs) inhibit insulin secretion and action in pancreatic beta cells. J. Biol. Chem. 2013, 288, 5682–5693.
- Cataldo Bascunan, L.R.; Lyons, C.; Bennet, H.; Artner, I.; Fex, M. Serotonergic regulation of insulin secretion. Acta Physiol. 2019, 225, e13101.
- Park, S.; Choi, S.B. Does fluoxetine administration influence insulin resistance in 90% pancreatectomized rats? Metabolism 2002, 51, 38–43.
More