1. Introduction
Bullous pemphigoid (BP) stands as the most common autoimmune blistering disease, presenting an estimated incidence ranging from 10 to 43 cases per million individuals per year
[1][2][1,2]. Remarkably, this disorder exhibits a distinct predilection for the elderly population, with escalating incidence beyond the age of 70 years old
[1][2][3][4][5][1,2,3,4,5]. According to a retrospective study conducted in the United Kingdom, the median age of BP onset was 80 years, underscoring the advanced age at which BP commonly manifests
[6].
The underlying mechanisms of BP remain largely unknown. However, it seems to rely upon the interaction between predisposing and triggering factors. Predisposing elements include genetic background, age and comorbidities such as neurological conditions. Eventually, the exposure to a specific trigger, such as drugs, physical factors, vaccines, infections or transplantations, holds the potential to induce or exacerbate BP
[7].
The diagnosis of BP is established through a combination of criteria, including clinical features, histopathological findings, positive direct immunofluorescence (DIF) and the detection of circulating IgG anti-basement membrane zone (BMZ) autoantibodies
[8].
The classical clinical manifestations of BP consist of tense bullae appearing on erythematous urticarial skin, primarily localized on the trunk and extremity flexures, as well as on the axillary and inguinal folds. Less frequently, bullae may appear on seemingly unaffected skin, a condition referred to as “non-inflammatory BP”. Regardless of the inflammatory background, BP is characterized by its intense associated pruritus
[1][8][1,8]. Mucosal involvement can be observed in up to 20% of BP patients, but it is mild and predominantly affects the oral cavity
[9]. Other bullous clinical variants include dyshidrosiform pemphioid, localized BP or lichen planus pemphigoides. Additionally, nonbullous presentations of BP encompass eczematous, urticarial and prurigo-like (pemphigoid nodularis) forms
[5][8][5,8].
Histopathological examination usually reveals subepidermal detachment containing eosinophils, neutrophils and fibrin, alongside a dermal inflammatory infiltrate. In non-bullous forms, skin biopsy shows eosinophilic spongiosis with an eosinophilic dermal inflammatory infiltrate, although these findings might be non-specific
[8]. Direct immunofluorescence (DIF) samples must be obtained from perilesional skin. The linear deposition of C3 and/or IgG along the BMZ in DIF displays the highest diagnostic sensitivity for BP (90.8%)
[10].
Indirect immunofluorescence (IIF) displays a linear IgG deposition along the dermoepidermal junction, which is shown to occur on the epidermal side of the split while using salt-split human skin as a substrate. Enzyme linked immunosorbent assay (ELISA) testing can detect and quantify serum levels of anti-BP180 and anti-BP230 autoantibodies, which are usually positive in BP with ranging sensitivity
[1]. A mosaic biochip designed to simultaneously detect multiple autoantibodies for the most common blistering diseases is commercially available. It has demonstrated high sensitivity and specificity, equivalent to each of its individual components, while streamlining the diagnostic process
[8][11][8,11].
2. Pathogenesis of Bullous Pemphigoid (BP)
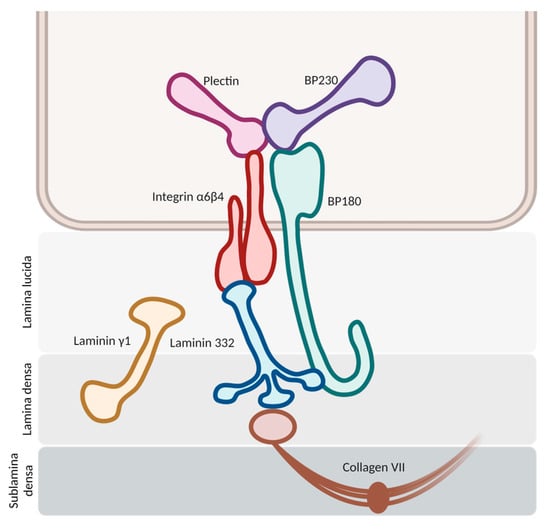
The pathogenesis initiates with the binding of autoantibodies against the hemidesmosomes in the basement membrane zone (BMZ) (
Figure 1). This binding activates multiple pathways, both complement-mediated and non-mediated, leading to the release of cytokines and proteases and the chemotaxis of neutrophils and eosinophils. Proteolytic cleavage at the BMZ induces dermal–epidermal separation and blister formation, with the subsequent dispersion of hemidesmosome-associated protein fragments. These fragments may interact with autoreactive lymphocytes, intensifying the inflammatory response
[1][5][1,5].
Figure 1. Schematic representation of hemidesmosomes in the basement membrane zone. The molecules of BP180 and BP230 stand as the main antigenic targets for autoantibody development in bullous pemphigoid.
2.1. Antigenic Targets
In bullous pemphigoid,
researchwe
rs usually find autoantibodies against two principal hemidesmosomal proteins: bullous pemphigoid antigen 2 (BPAg2) and bullous pemphigoid antigen 1 (BPAg1). BPAg2 is a 180 kilodalton transmembrane protein, also known as bullous pemphigoid 180 (BP180) or collagen XVII. BPAg1 is a 230 kilodalton intracellular hemidesmosomal protein, so it is also referred to as bullous pemphigoid antigen 230 (BP230)
[12].
2.1.1. BP180
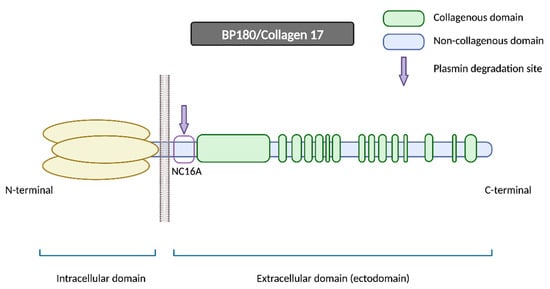
BP180 is a morphologically complex transmembrane protein (
Figure 2). It is composed of a globular intracellular domain in the amino-terminal and a large extracellular segment (or ectodomain) in the carboxyl-terminal that encompass the lamina lucida in the dermoepidermal junction and expands into the lamina densa
[13]. The ectodomain is composed of 15 collagenous domains interspersed with 16 non-collagenous (NC) domains, each designated in sequential order starting from the carboxyl-terminal (NC1, NC2 … NC16)
[14].
Figure 2. BP180/Collagen 17. BP180 is a transmembrane protein consisting of a globular intracellular domain at the amino-terminal and a large extracellular segment (or ectodomain) at the carboxyl-terminal. NC16A represents the juxtamembranous non-collagenous domain, and it contains the major pathogenic epitope for BP, in addition to being the primary site for plasmin and other serine proteases’ degradation. N-terminal: amino-terminal; C-terminal: carboxyl-terminal.
The NC16A domain consists of an extracellular juxtamembranous region and contains the major pathogenic epitope for BP. Thus, most common commercialized ELISAs for BP diagnosis use a recombinant NC16A protein to detect and quantify BP autoantibodies
[15] and 84–90% of BP sera react with the NC16A domain
[16][17][16,17]. It is important to note that anti-BP180 antibodies can also recognize other epitopes on BP180 beyond the NC16A domain, extending into the midportion or carboxyl-terminal regions of the ectodomain
[15]. Izumi et al. reported that these cases with non-NC16A anti-BP180 antibodies displayed a non-inflammatory phenotype with less erythema and a diminished eosinophilic infiltrate and were more likely to respond to corticosteroid treatment
[18].
One of the hypotheses that has been put forth to explain the disease revolves around impaired proteolytic degradation. The physiological shedding of BP180 by serine proteases, including plasmin, exposes new antigens and generates neoepitopes, which could serve as targets for blister-inducing antibodies. The principal site of degradation for the BP180 molecule is the juxtamembranous domain NC16A, aligning with the major pathogenic epitope of BP
[18][19][18,19]. Other proteinases, such as A Disintegrin and Metalloproteases (ADAMs) and Granzyme B (GzmB), might also contribute to the generation of neoepitopes and the onset of BP through BP180 cleavage. In fact, GzmB expression is upregulated with age, which could help to explain its role in this age-related autoimmune blistering disorder
[20].
BP180 has also been demonstrated to be present in extracutaneous tissues, such as in various neuroanatomical regions in the brain
[21], and as a component of the glomerular filtration barrier in the kidneys
[22]. However, its precise function and potential role in neurodegenerative disorders or renal diseases remains to be elucidated
[21][22][23][21,22,23].
2.1.2. BP230
BP230 is an intracellular component of hemidesmosomes and is part of the plakine family. Anti-BP230 antibodies mainly bind to the globular carboxyl-terminal domain and are detected in approximately 60–70% of BP serum samples
[24]. Given the fully intracellular location of BP230, the accessibility of autoantibodies to this antigen is potentially limited. Consequently, it remains uncertain whether they exert a pathogenic role in BP or merely appear as a secondary event linked to keratinocyte injury (occurring as byproducts of epitope spreading associated with disease extension)
[16].
Nonetheless, anti-BP230 antibodies have been associated with the appearance of non-bullous pemphigoid
[25], whereas its absence may correlate with mucosal involvement in BP patients
[9]. As a result, anti-BP230 antibodies might contribute to some extent in the development of BP; however, the precise mechanism and significance remain unclear.
2.2. Hypothesis of Blister Formation
It is well accepted that bullous pemphigoid arises from a loss of immune tolerance, resulting in the production of autoantibodies against BP180 and BP230. These antibodies trigger an inflammatory reaction, attracting numerous neutrophils, eosinophils and mast cells, which migrate to the dermis and release a wide range of cytokines and proteases, responsible for dermoepidermal cleavage and blister formation
[26].
Until the last decade, complement was believed to be a prerequisite for blister formation by autoantibodies. Complement components are present along the dermoepidermal junction in patients with BP, as demonstrated with direct immunofluorescence (DIF), which shows linear C3 deposition in 83–84% of BP cases
[27][28][27,28]. Complement proteins are present also in the blister fluid of BP patients
[29]. Furthermore, complement activation by autoantibodies may correlate with disease activity, as demonstrated in laboratory and clinical studies
[28][30][28,30]. All this evidence suggests a potential pathogenic role of the complement system in BP development.
Nonetheless, more recent studies have questioned its major pathogenic role in bullous pemphigoid, proposing the existence of complement-independent mechanisms mediating blister formation
[28][30][28,30]. In animal models, Ujiie et al. demonstrated that the passive transfer of BP autoantibodies induced blister formation in C3-deficient humanized mice, despite not being able to activate the complement cascade
[31]. Furthermore, it should be noted that if DIF shows a C3 deposition in 83–84% of patients, then 16–17% of BP patients do not present this complement protein along the dermoepidermal junction and thus they might not be mediated by the complement system
[27][28][27,28]. In these cases, IgG4 antibodies are the dominant IgG subclass, which are not able to activate the complement cascade
[32].
2.2.1. Complement-Dependent Immune Response
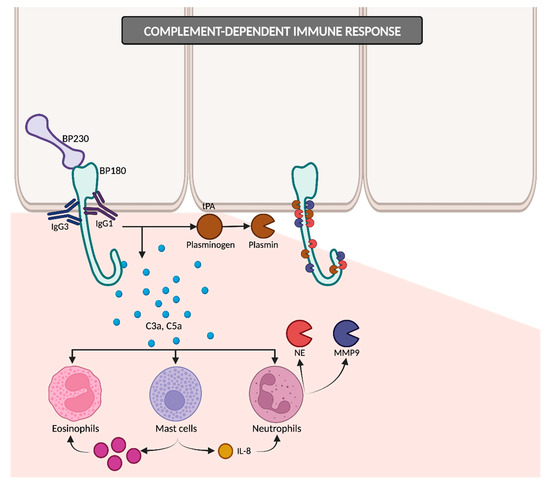
The IgG1 and IgG3 antibodies bind to BP180, consequently initiating the activation of the complement cascade (
Figure 3). The resulting anaphylatoxins C3a and C5a induce the chemotaxis and degranulation of neutrophils, eosinophils and mast cells. Neutrophils release proteolytic enzymes, including neutrophil elastase (NE) and matrix metalloproteinase 9 (MMP9), leading to the degradation of BP180 and subsequently weakening basal cells’ adhesion to the basement membrane zone (BMZ). Simultaneously, mast cells secrete IL-8, which amplifies the neutrophilic infiltration, and numerous proinflammatory cytokines that recruit additional eosinophils. Upon reaching the BMZ, migrated eosinophils discharge their granule proteins, culminating in subepidermal blistering
[12][33][12,33]. In addition to NE and MMP9, other proteases may potentially contribute to dermal–epidermal cleavage, as evidenced by studies detecting plasmin in BP blister fluid
[29][34][29,34]. The binding of IgG to BP180 on keratinocytes could induce the liberation of tissue-type plasminogen activator (tPA), thereby catalyzing the conversion of plasminogen into active plasmin
[18][34][18,34].
Figure 3. Complement-dependent immune response. IgG1 and IgG3 antibodies bind to BP180 and initiate the complement cascade, leading to eosinophils, mast cells and neutrophils’ recruitment and subsequent degranulation. IgG binding on keratinocytes may also trigger tPA release, converting plasminogen into plasmin. Plasmin, along with NE and MMP9, promotes dermal–epidermal cleavage. tPA: tissue-type plasminogen activator; NE: neutrophil elastase; MMP9: matrix metalloproteinase 9.
2.2.2. Complement-Independent Immune Response
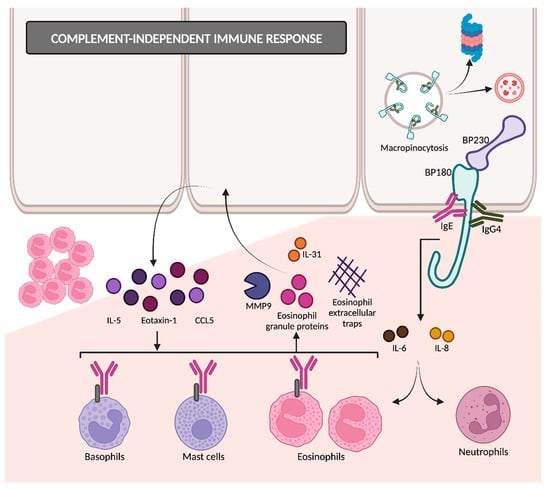
The binding of autoantibodies to BP180 in hemidesmosomes results in the internalization of BP180 into basal keratinocytes (
Figure 4)
[35], so the adhesive strength of the dermoepidermal junction decreases. This appears to be an early event in disease pathogenesis, followed by an inflammatory response that finally causes dermoepidermal separation
[26][36][26,36]. BP180 internalization occurs through the micropinocytosis pathway
[36]. Afterwards, it remains unclear whether they are degraded in lysosomes (as macropinosomes usually do) or if it is mediated by ubiquitylation and proteasomal degradation
[14].
Figure 4. Complement-independent immune response. IgG4 and IgE are involved in BP pathogenesis through complement-independent mechanisms. Autoantibody binding to BP180 results in its internalization through the micropinocytosis pathway. The interaction between BP180 and autoantibodies triggers the release of several cytokines, ultimately attracting eosinophils and proteases that contribute to dermal–epidermal separation. MMP9: matrix metalloproteinase 9; CCL5: chemokine ligand 5.
Following the interaction between anti-BP180 antibodies and BP180 ectodomain, keratinocytes release proinflammatory cytokines such as IL-6 and IL-8
[37], possibly mediated by the upregulation of NF-kappa beta and STAT3
[38]. These cytokines and chemokines attract eosinophils and neutrophils, responsible for the inflammatory reaction
[39].
IgG4 has a very limited ability to fix complements and has been reported by some studies as the predominant subclass of autoantibodies in BP, followed by IgG1 and IgG3
[40][41][40,41]. The dominance of IgG4 is more evident in the early stages of BP, suggesting that IgG4 may play a pathogenic role primarily in the initiation of the immune response
[41].
The balance between the contributions of complement-dependent IgG1 and complement-independent IgG4 might explain the clinical diversity that
researchwe
rs can find in BP. Certain authors support that C3-positive pemphigoids in DIF are mediated by IgG1/IgG3 and IgG4, and they would present as the classic BP with urticarial rash and worse clinical severity. On the other hand, C3-negative pemphigoids are IgG4-dominant and tend to have a non-inflammatory phenotype with milder severity
[39].
Another immunoglobulin with little ability to activate complements is IgE, which is increasingly being linked to the pathogenesis of BP. Anti-BP180 IgE autoantibodies are detected in the majority of BP sera and are correlated with disease activity
[42][43][42,43]. BP180–IgE complexes adhere to the keratinocyte basement membrane and bind with the FcεR1 receptors present on eosinophils, mast cells and basophils. This interaction triggers the release of proteases such as MMP9, eosinophil granule proteins and eosinophil extracellular traps. MMP9 degrades BP180, thereby contributing to dermal–epidermal separation. Eosinophils also secrete interleukin 31, which is directly related to pruritus in BP. In response to eosinophil granule proteins, keratinocytes release cytokines such as IL-5, eotaxin-1 and chemokine ligand 5 (CCL5). This cyclical process amplifies tissue eosinophilia and promotes eosinophilic spongiosis
[26][27][44][26,27,44]. These facts support the relevance of complement-independent Th2-mediated pathways in the pathogenesis of BP.
Hence, it is plausible that both complement-dependent and independent mechanisms play a collaborative role in triggering and perpetuating bullous pemphigoid
[45].
2.3. Breakdown of Self-Tolerance
The fundamental initial process in the development of bullous pemphigoid is the generation of autoantibodies targeting hemidesmosomal proteins. FoxP3+ regulatory T cells (FoxP3+ Treg) represent the pivotal cell population for self-tolerance maintenance, since they are responsible for suppressing excessive autoantibody production
[16]. However, the scientific literature exhibits contradictory results in this regard. Some authors have documented decreased FoxP3+ Treg cells among BP patients
[46][47][46,47], whereas other authors have identified a substantial increase
[48]. These differences may stem from a selection bias associated with Treg markers. Specifically, Muramatsu et al. reported that total Treg cells are increased in classic BP patients before treatment, possibly secondary to the inflammatory background, but significantly decrease after corticosteroid treatment. This finding can be attributed to the inhibition of IL-2 by corticosteroids, which is required for the maintenance of Treg cells. Alternatively, corticosteroid treatment might suppress autoreactive T cells and therefore effector Treg cells would consequently decrease as they are no longer needed
[49].
Nevertheless, the dysfunction of Treg cells has been identified in BP
[50]. This malfunction can result in the suppression of self-tolerance and subsequently the formation of autoreactive T helper 2 (Th2) lymphocytes mediated by STAT6. Autoreactive Th2 cells are able to activate and sensitize B cells and generate antibodies against self-components
[16].
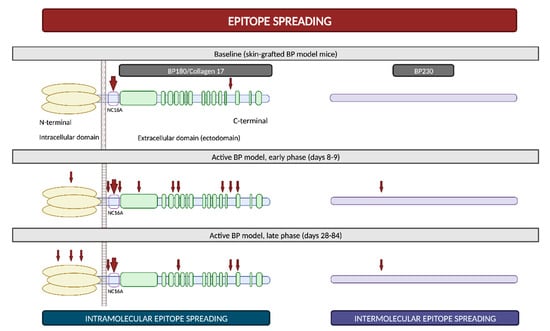
2.4. Epitope Spreading
Epitope spreading (ES) is a phenomenon in which the immune responses of T and/or B cells extend from the original dominant epitope to other secondary epitopes as time progresses. These new epitopes may be located on the same autoantigen (intramolecular epitope spreading) or on different antigens within the same anatomical site
[51].
It is widely recognized that ES is a frequent event in the development of BP. In vivo studies using murine models have demonstrated that IgG antibodies targeting BP180 initially react to epitopes situated within the ectodomain and, subsequently, they react to other extracellular and intracellular domains over time (
Figure 5)
[52][53][52,53]. However, this immunological reaction is not solely confined to antigens localized within BP180; rather, it progressively spreads over time to other molecules, including BP230
[53]. Furthermore, in a prospective multicenter study, ES was observed in 49% of patients following a 1-year observational period. These events exhibited a distinct propensity to occur during the early stages of the disease
[54]. All these findings suggest that NC16A recognition in the BP180 ectodomain is an early event, succeeded by intra- and intermolecular ES events. These sequential occurrences collaboratively mold the individual course of each patient with BP
[51].
Figure 5. Epitope spreading. Epitope spreading in murine models according to the research conducted by Ujiie et al. Figure adapted from Ujiie et al. (2019)
[53].
The concept of epitope spreading has been suggested as an explanation for those cases in which BP develops in the setting of other diseases
[53]. For example, the basement membrane zone disruption in oral lichen planus might expose hemidesmosome proteins and then trigger the autoimmune humoral response responsible for lichen planus pemphigoides
[55]. BP may also develop after radiation therapy, possibly through the exposure of BMZ antigens during the course of the treatment
[56]. Finally, ES from brain BP180 due to neurologic damage has also been proposed to partially explain the relationship between BP and certain neurocognitive diseases
[57]. Although BP180 is diffusely expressed within the central nervous system, a recent study has revealed that it is not expressed in the hippocampus, which is the main area affected in neurocognitive disorders
[23]. This underscores the need for future research to elucidate the intricate connection between neurological disorders and BP.
3. General Aspects of Drug-Associated Bullous Pemphigoid (DABP)
3.1. Drugs Related to DABP
The first case of drug-associated bullous pemphigoid (DABP) was reported in an 11-year-old patient receiving treatment with salicylazosulphapyridine
[58]. Subsequently, a wide range of drugs have been linked to the pathogenesis of this disease.
According to the Naranjo Adverse Reaction Probability Scale
[59] and the Karch–Lasagna algorithm
[60], most bullous pemphigoid cases could be categorized as having a “probable” association with a drug regarding the temporal relationship, the available literature and the absence of alternative causes. However, while these scales are useful in assessing general drug reactions, their significance appears diminished when applied to the identification of potential triggers in drug-associated bullous pemphigoid. In contrast, Tan et al. proposed specific criteria to consider a drug as a potential trigger for BP. These criteria include the drug’s initiation within the preceding year, a treatment duration of more than 2 weeks and drug continuation until at least 1 month before the diagnosis of BP
[61].
Medications most frequently linked to BP include dipeptidyl peptidase 4 inhibitors (DPP4i), diuretics, neuroleptics, antibiotics, monoclonal antibodies against anti-tumor necrosis factor (TNF)-α, immune checkpoint inhibitors targeting programmed cell death protein 1 (PD-1) and its ligand (PD-L1), non-steroidal anti-inflammatory drugs (NSAID) and antihypertensive drugs. However, the list is exponentially growing (
Table 1). BP has even been reported to develop after the application of certain topical drugs, inducing some form of “contact pemphigoid”
[62]. However, the potential of topical agents to trigger BP remains controversial, as direct associations are not well established in most cases
[63].
Table 1. List of drugs that have been linked to bullous pemphigoid development according to the scientific literature.
Verheyden et al. conducted a systematic review of drug-associated bullous pemphigoid and consequently developed a diagrammatic summary of the strength of supporting evidence for each drug. Within this framework, the evidence was stronger for DPP4i, followed by immune checkpoint inhibitors PD-1/PD-L1, loop diuretics, penicillins, NSAIDs, thiazides and psoralens with ultraviolet A phototherapy
[4]. Additionally, Liu et al. recently published a meta-analysis of case–control studies, in which they found a significant association between BP and the prior use of DPP4i (odds ratio [OR] 1.92), aldosterone antagonists (OR 1.75), anticholinergics (OR 3.12) and dopaminergic medications (OR 2.03)
[64].
Nonetheless, the majority of these associations are predominantly drawn from case reports, relying on factors such as temporal correlation or similarity to previously reported cases. As a result, the levels of evidence for most of the suspected medications are low due to the absence of controlled studies
[64]. Furthermore, these clinical associations are subject to various confounding elements, including the prevalent polypharmacy among elderly individuals and the common use of over-the-counter drugs that are seldom reported to healthcare professionals. Unfortunately, ethical and safety concerns make it infeasible to rechallenge patients in order to definitively confirm the presumed link between BP and drug exposure
[4][63][4,63].
As ongoing research continues to unravel the pathogenesis and natural history of DABP, clinicians should be aware of this association in order to identify and treat potential cases of DABP early on
[4][63][4,63]. Drug discontinuation in DABP might lead to a reduction in the need for immunosuppression and a better prognosis when compared to missed DABP
[65].
3.2. Pathogenic Mechanisms
Increasing interest is being directed towards the research of DABP pathogenesis, yet a precise understanding of the underlying mechanisms is still lacking. Drugs are thought to act as triggering factors in patients with an underlying genetic susceptibility. Various studies have suggested a potential correlation between DABP and specific major histocompatibility complex (MHC) class II alleles, since they could facilitate the presentation of BMZ autoantigens to T cells
[4][7][4,7].
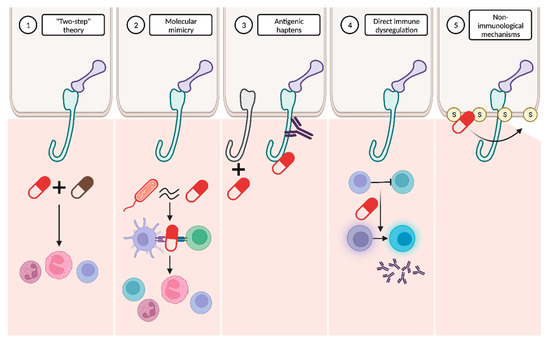
It has been hypothesized that the pathogenesis of DABP might be explained by the interaction of several mechanisms (Figure 6).
Figure 6. Proposed mechanisms to explain drug-associated bullous pemphigoid pathogenesis. (1) The “two-step” theory proposes that the interaction between two drugs may be necessary to initiate and amplify the immune response. (2) The molecular mimicry hypothesis suggests that the molecular similarity between certain drugs and microorganisms could trigger an immune response against the drugs. (3) Other drugs may act as antigenic haptens, modifying the antigenic properties of specific proteins. (4) Certain drugs may directly induce immune dysregulation. (5) Non-immunological mechanisms, involving interaction with sulfhydryl groups present in the dermoepidermal junction, are also plausible.
-
“Two-step” theory: the interplay between two drugs with analogous molecular structures and their interaction with the immune system might represent the first and second “hits” required to initiate and amplify the immune response
[4][7][63][4,7,63].
-
Molecular mimicry: many drugs bind to RNA and proteins in a way that closely resembles the interaction pattern observed with viruses. This similarity raises the possibility that these drugs might be erroneously recognized as microbial antigens. The immune system’s misidentification of drugs in predisposed individuals could result in the activation of CD4+ T cells and the subsequent initiation of the autoimmune cascade
[7][7[63],63].
-
Antigenic haptens: some drugs may have the ability to function as antigenic haptens that can bind to and modify protein molecules within the lamina lucida of the BMZ. Such interactions might induce the modification of their antigenic properties, thereby acting as neoantigens. Alternatively, this phenomenon could lead to the exposure of a previously hidden antigenic site, supporting the drug-triggering epitope spreading theory
[4][7][63][66][4,7,63,66].
-
Direct immune dysregulation: drugs may cause immune reorganization, disrupting the endogenous regulatory processes that prevent the development of several diseases. Alterations in T-regulatory cell functions may suppress “forbidden” B cell clones and then result in the release of autoantibodies against the BMZ
[63][66][63,66].
-
Non-immunological mechanisms: thiol-containing drugs may directly interact with the sulfhydryl groups present in the BMZ proteins and subsequently disrupt the dermoepidermal junction without the involvement of immunological mechanisms. However, this dermoepidermal cleavage may also expose new, hidden antigenic sites
[7][63][7,63].
Furthermore, Verheyden et al.
[4] recently proposed that drugs related to BP may be categorized according to their chemical structure as thiol-based, phenol-based and non-thiol non-phenol-based drugs.
-
Thiol-based drugs: they might induce BP acting as haptens or directly disrupting the dermoepidermal junction, as previously described. Moreover, penicillamine, a specific thiol-based drug, could decrease the activity of T-regulatory cells
[62]. Many drugs, such as furosemide, hydrochlorothiazide, spironolactone, penicillins or sulfasalazine, contain sulfur atoms within their molecules, yet not as part of a thiol group. However, it is hypothesized that they may be able to form thiol groups during their metabolism, thereby inducing BP through a similar mechanism to thiol-based drugs
[67].
-
Phenol-based drugs: these medications incorporate a phenyl group in their molecular structure and are thought to interfere with the integrity of the BMZ, consequently revealing hidden epitopes. Examples of these phenol drugs are non-steroidal anti-inflammatory drugs (NSAID), cephalosporins, angiotensin II receptor blockers (ARB) and selective serotonin reuptake inhibitors (SSRI).
-
Non-thiol non-phenol-based drugs: the number of these drugs is continuously growing, although the precise underlying mechanisms remain largely undefined.
3.3. General Differences between DABP and Idiopathic BP
3.3.1. Clinical Differences
Patients diagnosed with DABP are often younger than those affected by the idiopathic form
[4][63][4,63]. In contrast to classic BP, the clinical manifestations of DABP may be more heterogenous, resembling other conditions like erythema multiforme or pemphigus, which often delays the diagnosis
[4][68][4,68]. Lesions typically manifest as tense bullae on seemingly normal skin or, more infrequently, on an erythematous or urticarial base
[63].
The natural course of DABP remains somewhat uncertain, although there have been recognized two variants based on their clinical history. The first is an acute, self-limited form characterized by definitive resolution upon discontinuation of the suspected drug. This form can be genuinely categorized as a drug reaction (true drug-induced bullous pemphigoid). Conversely, the second form presents a chronic and severe course, similar to classic bullous pemphigoid. It can persist even after the suspected drug is withdrawn and may require a prolonged treatment (drug-triggered bullous pemphigoid)
[66].
3.3.2. Histological and Laboratory Differences
Despite all the extensive research conducted in bullous pemphigoid, no specific antigens for DABP have been identified. Therefore, it is believed that the autoantigens involved might align with those identified in idiopathic BP. Direct and indirect immunofluorescence typically exhibit similar patterns to those of idiopathic BP
[4][63][4,63].
The typical histological findings in DABP encompass the presence of intraepidermal vesicles, necrotic keratinocytes and a prominent eosinophilic infiltrate, with occasional thrombus formation. However, idiopathic BP typically lacks intraepidermal vesicles, necrotic keratinocytes and thrombi, and the eosinophilic infiltrate is usually milder. Marked eosinophilia in serum is frequently observed in DABP cases
[4][63][4,63].