Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Juan Duarte.
Systemic lupus erythematosus (SLE) is a multifactorial disorder with contributions from hormones, genetics, and the environment, predominantly affecting young women. Cardiovascular disease is the primary cause of mortality in SLE, and hypertension is more prevalent among SLE patients.
- systemic lupus erythematosus
- endothelial dysfunction
- hypertension
1. Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by multisystemic inflammation and organ damage manifestations that affect the skin, joints, kidneys, heart, lungs, blood, and the central nervous system [1]. Autoantibodies targeting the cell nucleus are present in 99% of SLE patients, and autoantibodies specific to double-stranded DNA (anti-dsDNA) have been identified in more than 70% of patients [2]. Even though the presence of anti-dsDNA is predictive in 95% of SLE cases, a more extensive immune dysregulation is involved in the etiopathogenesis of SLE, although the exact cause of SLE remains unclear [3]. In a study by Wang et al., mitochondrial DNA (mtDNA) was detected in neutrophils with extracellular traps (NETs) [4]. The researchers observed elevated levels of anti-mtDNA antibodies in SLE patients compared to control subjects, which were significantly correlated with disease activity. Moreover, the presence of anti-mtDNA antibodies was disproportionately associated with lupus nephritis and showed a stronger correlation with the lupus nephritis activity index compared to anti-dsDNA levels [4].
SLE is a complex disorder influenced by a combination of hormonal, genetic, and environmental factors, primarily affecting young women (with a female-to-male ratio of 9 to 1) and typically manifesting during the reproductive years. The leading cause of mortality in SLE is cardiovascular disease (CVD), which can be attributed to a combination of risk factors such as hypertension, dyslipidemia, and a prothrombotic state. Increased atherosclerosis has already been demonstrated in SLE [5]. SLE has been linked to a proatherogenic lipid profile that includes elevated total cholesterol, triglycerides, low-density lipoprotein cholesterol (LDL-C), and high-density lipoprotein cholesterol (HDL-C) [6]. The most common abnormality seen in SLE patients is a decrease in HDL-C. The prevalence of dyslipidemia was 36% at the time of diagnosis and 60% at 3-year follow-up in a cohort of 918 SLE patients [7,8][7][8]. In this study, the prevalence of other traditional risk factors, such current smoking and diabetes, at enrollment was 13.7% and 3.4%, respectively, which was also increased through the follow-up [7,8][7][8]. Furthermore, metabolic syndrome is recognized as a proinflammatory state that may contribute to premature atherosclerosis and diabetes development in SLE patients, increasing their CVD risk. Metabolic syndrome is more common in SLE patients [9]. A recent systematic review and meta-analysis looked at the risk of CV events and CV risk factors in adult SLE patients. When compared to adults without SLE, the relative risk (RR) of hypertension was higher at 2.7, whereas the RRs of diabetes and metabolic syndrome were elevated but not statistically significant [10]. However, while traditional Framingham risk factors may contribute to CVD pathogenesis in SLE patients, they cannot fully explain the increased CVD risk in SLE patients [11]. Indeed, certain SLE-specific factors may play a role in CVD onset and progression in SLE patients. Additionally, another contributing factor to cardiovascular disease in SLE is the presence of elevated antiphospholipid antibodies, which can directly induce pro-inflammatory and prothrombotic effects on the endothelium and disrupt coagulation by inhibiting annexin A5, thereby negating its antithrombotic and protective effects [12].
Zhao M et al. reported that 71.9% of SLE patients had hypertension, and a significant portion of them, specifically 74.4%, were either undertreated or not treated at all [13]. Despite the high prevalence of hypertension among SLE patients, current hypertension management guidelines do not address the specific needs of individuals with autoimmune disorders like SLE. This has led healthcare providers to rely on recommendations designed for the general population, lacking data from comprehensive clinical trials within the SLE patient group [14]. Consequently, many patients with this autoimmune disease do not receive the appropriate antihypertensive medications they may require [15].
2. Systemic Lupus Erythematosus and Arterial Hypertension
Hypertension stands as the leading risk factor for the progression of renal, vascular, and cardiac diseases in the general population [19][16], which is exacerbated by immune-mediated mechanisms in SLE patients. Multiple studies highlight the elevated prevalence of hypertension in women with SLE. Women aged 35 to 44 with SLE are 50 times more likely to experience a cardiac event, such as infarction or angina, than individuals of the same age without the condition [12,20][12][17]. Nonetheless, there is a scarcity of mechanistic studies on hypertension in SLE, and the precise underlying mechanisms remain unknown [21][18]. The renal pathological mechanisms related to autoimmune-induced hypertension remain incompletely understood. It is established that a loss of self-tolerance results in the production of autoantibodies. These autoantibodies form complexes, which deposit into tissues such as the kidneys, leading to the activation of other immune cells and the complement system. This, in turn, triggers the local secretion of inflammatory mediators, promoting chronic renal inflammation and oxidative stress [22][19]. These processes can potentially disrupt fluid and electrolyte balance in the kidneys or lead to renal vascular dysfunction, subsequently causing hypertension [23][20]. Various animal models of SLE have been employed to investigate the genetic and immunological mechanisms contributing to this autoimmune disorder. The NZBWF1 mouse model, utilized for over four decades, closely resembles human lupus nephritis. These mice exhibit features like immunocomplex deposition in glomeruli, high plasma levels of anti-dsDNA, albuminuria, and hypertension between 25–30 weeks of age [24][21]. In both female humans with SLE and the NZBWF1 mouse model, lupus nephritis precedes hypertension. This provides an opportunity to study the contributing factors to hypertension in the context of chronic renal inflammation [25][22]. However, like humans, it exists a disparity between nephritis and blood pressure in animal models of SLE. For instance, MRL/lpr, BXSB, and NZBWF1 mice develop glomerulonephritis, but only the NZBWF1 mouse model develops hypertension [26][23]. Hence, the NZBWF1 model is especially valuable for understanding the pathophysiology of hypertension in SLE. Several factors contribute to hypertension in this model, including altered renal hemodynamics, endothelial dysfunction, changes in the inflammatory cytokine profile, oxidative stress, dysfunction in the adaptive immune system, and sex hormones [21][18]. However, it is important to note that hypertension in this lupus mouse model is not salt-sensitive [27][24]. This precludes the use of diuretics as a treatment option. Nevertheless, angiotensin-converting enzyme inhibitor drugs (ACEIs) like captopril and enalapril delay the onset of renal damage and reduce blood pressure, while cyclophosphamide treatment does not have effect in blood pressure [28,29][25][26]. Moreover, captopril reduced chronic renal lesions by reducing TGF-beta expression in the kidneys but had no effect on autoantibody production [30][27]. Recently, a murine model of lupus induced by topical administration of imiquimod, a toll-like receptor (TLR) 7 agonist, in the ears of BALB/c mice has been established [31][28]. These mice develop splenomegaly, autoantibody production, and glomerulonephritis with immunocomplex deposition within 4–6 weeks. Furthermore, they exhibit elevated levels of type I interferon, a result of TLR7 activation. It is worth noting that gain-of-function variants of TLR7 have been linked to lupus nephritis [32][29]. This model has also shown signs of endothelial dysfunction [33][30] and hypertension [18][31], indicating that TLR7 endosomal receptor activation, triggered by self-antigens (ds-DNA and ssRNA), may contribute to lupus-associated vasculopathy. Additionally, wresearchers have observed an approximately 2.5-fold increase in TLR7 expression in the vascular wall (unpublished data) in hypertensive NZBWF1 mice. However, the relevance of this finding to the occurrence of endothelial dysfunction in this model remains unknown. In another murine model induced by pristane to mimic SLE, which is widely used to evaluate potential therapeutic agents resembling human idiopathic lupus syndrome [34][32], hypertension, chronic inflammation, and endothelial dysfunction were also observed [35][33]. There is limited evidence regarding the impact of first line SLE therapies on blood pressure. Corticosteroids, the most commonly used agents for all autoimmune diseases, have been associated with hypertension in SLE patients. Furthermore, women who had used prednisone for a longer period and had a higher cumulative dose of prednisone, as well as those who had previously had a coronary event, were more likely to have plaque [36][34]. On the other hand, mycophenolate mofetil (MMF) and hydroxychloroquine have demonstrated beneficial effects on blood pressure, independently of additional antihypertensive treatments and existing renal disease [37][35]. Several mechanisms by which hydroxychloroquine may protect against vascular injury have been proposed in mouse studies, including the reduction of vascular oxidative stress via antioxidant action in a mouse model of SLE [17,38][36][37] and the regulation of endothelial nitric oxide synthase in a mouse model of antiphospholipid syndrome [39][38]. Novel immunomodulatory drugs like belimumab and anifrolumab offer promising options for regulating immune system activation in SLE, but their effects on renal inflammation and hypertension in SLE have received limited investigation. Other new immunomodulatory drugs include JAK inhibitors (jakinibs), which inhibit the downstream cellular effects of various cytokines, including type I IFNs and cytokines involved in neutrophil development and function. Jakinib tofacitinib was shown to be effective in reversing lupus symptoms and vascular dysfunction in mice, as well as inhibiting netosis [40][39]. Short-term use of tofacitinib in SLE patients with mild–moderate disease activity was safe and improved arterial stiffness [41][40]; however, long-term clinical studies are needed to determine whether this treatment confers benefits for SLE patients addressing CV morbidity and mortality, especially given a reported link between thrombosis and JAK inhibition [42][41].2.1. Role of Endothelial Function in SLE Hypertension
A substantial number of individuals with SLE exhibit signs of subclinical vascular disease that precede the onset of atherosclerosis. These subclinical changes include the development of endothelial dysfunction (with preserved vascular smooth muscle function) [43][42], arterial wall thickening, and coronary perfusion abnormalities [44][43]. Multiple studies emphasize the prominent impact on the endothelium in SLE. A meta-analysis of 25 case-control studies encompassing 1313 SLE patients and 1012 healthy controls, utilizing a random effects model, revealed that SLE patients exhibited lower brachial artery endothelium-dependent flow-mediated dilation compared to healthy controls [45][44]. Notably, diabetes mellitus, renal disease, and diastolic hypertension are significant contributors to endothelial dysfunction in SLE patients [45][44]. However, whether endothelial dysfunction plays a causal role in SLE-associated hypertension or is merely an associated condition remains uncertain. Possible mechanisms underlying endothelial dysfunction in SLE encompass netosis, endotoxemia, and an imbalance between pro-inflammatory and anti-inflammatory cytokines, leading to reduced nitric oxide (NO) bioavailability, endothelial leakage, and impaired endothelial repair (Figure 1). The decreased vasodilator response to acetylcholine in NZBWF1 mice is observed before the onset of proteinuria and elevated blood pressure, suggesting that these early vascular function changes may contribute to the development of hypertension in SLE [46][45]. The specific causes of endothelial dysfunction in the NZBWF1 model are not yet completely understood. Oxidative stress and proinflammatory cytokines have been proposed as potential factors contributing to hypertensive endothelial dysfunction in SLE.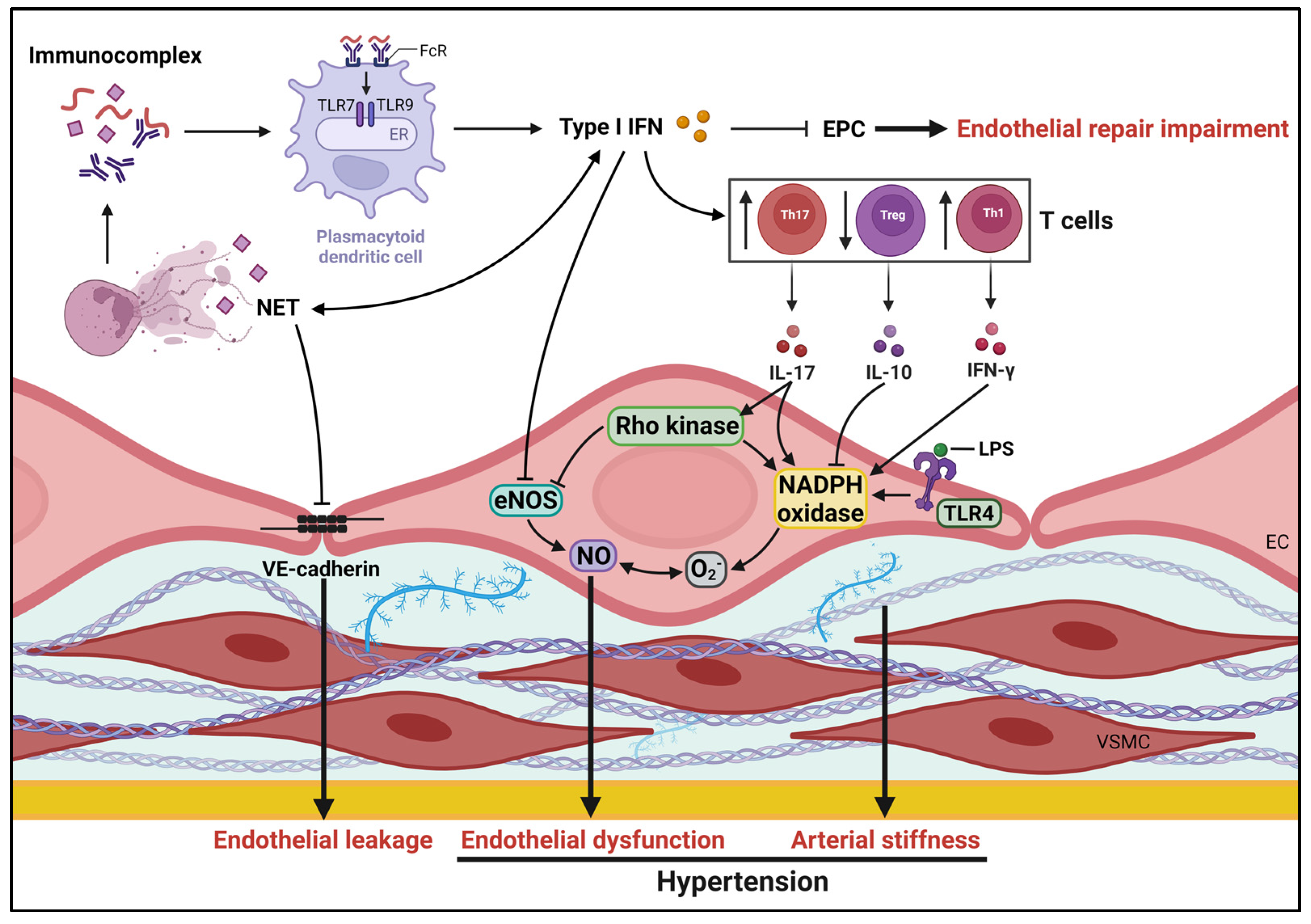
Figure 1. Mechanisms potentially underlying pathogenesis of hypertension in SLE. Several SLE-specific factors, such as impairment of endothelial repair, endothelial dysfunction and arterial stiffness influence vascular health, contributing to development of hypertension. eNOS, endothelial nitric oxide synthase; ECs, endothelial cells; EPCs, endothelial progenitor cells; ER, endoplasmic reticulum; FcRs, Fc receptors; IFN, interferon; IL, interleukin; NETs, neutrophil extracellular traps; NO, nitric oxide; LPS, lipopolysaccharide; TLR, Toll-like receptors; VSMCs, vascular smooth muscle cells.
2.2. Immune System and Hypertension in SLE
It is now commonly accepted that aberrant immune system activation is linked to the development of hypertension, both in humans and experimental models. In fact, there have been numerous genetic polymorphisms associated with hypertension described that exert effects on the immune response [63][62]. Non-specific immunosuppressive therapies, such as cyclophosphamide, have been proven in experimental models of spontaneous non-autoimmune hypertension to not only prevent the development of hypertension but also to lower blood pressure in animals with established hypertension [64][63]. Similarly, MMF, an immunosuppressive therapy frequently used in SLE patients to suppress T and B lymphocyte proliferation, has been effective in attenuating the development of hypertension in various experimental models and in humans [65,66,67][64][65][66]. Several studies have found a link between circulating autoantibodies, which are seen in systemic autoimmune diseases, and essential hypertension in humans. SLE is a chronic autoimmune disorder characterized by the hyperreactivity of B and T lymphocytes due to a loss of tolerance to self-antigens, resulting in the production of pathogenic autoantibodies, particularly against nuclear components. However, it remains unclear whether the autoantibodies produced in SLE contribute mechanistically to the development of hypertension in these patients. The immunization process leading to the abnormal production of autoantibodies in SLE is partly attributed to the delayed clearance of apoptotic cells. OurThe research has demonstrated that the activation of peroxisome proliferator-activated receptors β/δ (PPARβ/δ) plays a crucial role in the proper clearance of deceased cells by macrophages in the NZBWF1 model, which has been associated with a reduction in elevated blood pressure [47][46]. Animal models of SLE are critical in understanding the relationship between autoantibodies and hypertension. As previously stated, the NZBWF1 mouse model closely resembles key characteristics of clinical SLE, including autoantibody generation, immune-complex-mediated kidney damage, and hypertension. Recent studies have shown that long-term B cell depletion using anti-CD20 successfully reduced autoantibody synthesis and prevented the onset of hypertension in SLE mice [68][67]. Furthermore, continuous administration of the immunosuppressive medication MMF selectively lowered B cells while preventing the development of hypertension [69][68]. These data unequivocally indicate a relationship between B cells, autoantibodies, and the onset of hypertension. However, it is important to highlight that these treatments were only beneficial when started before the commencement of the disease. Similarly, treatment efforts targeting B cells in humans, such as anti-CD20 (rituximab), have had minimal effectiveness in large controlled clinical studies [70][69]. It has been suggested that the low efficacy is due in part to the persistence of plasma cells, which are not the target of B cell treatments. Plasma cells take up residence in the bone marrow and spleen for extended periods, ranging from months to years, and are primarily responsible for producing serum immunoglobulins, including SLE autoantibodies. The depletion of plasma cells using the proteasome inhibitor bortezomib has been demonstrated to diminish autoantibody production and alleviate hypertension in female NZBWF1 mice. Collectively, these findings indicate that the mechanistic link between autoantibody production and autoimmune-associated hypertension involves plasma cells [71][70]. Autoantibody production and the clinical severity of lupus in NZBWF1 mice prone to the disease are contingent on the assistance of CD4+ T cells [72][71]. Activated B cells migrate to areas rich in T cells within secondary lymphoid organs, where they recruit antigen-bound T cells. Subsequently, B cell differentiation occurs, either extrafollicularly into plasmablasts, with limited antibody production capacity, or by transitioning along the follicular pathway, forming germinal centers (GCs). Within GCs, follicular T-helper (Tfh) cells facilitate somatic hypermutation and isotype switching of B cells, culminating in the generation of long-lived plasma cells capable of producing high-affinity antibodies [73,74][72][73]. In a murine model of SLE, a connection between increased Tfh cell numbers and the development of autoimmunity has been established [75[74][75],76], suggesting that anomalies in the positive selection of Tfh cells could lead to systemic autoimmunity. Tregs also play a role in regulating the production of lupus-associated antibodies, including anti-dsDNA [77][76]. A substantial inverse correlation has been observed between Tregs and anti-dsDNA levels [78][77]. Although the precise role of Tregs in the pathogenesis of SLE remains to be fully elucidated, expanding Tregs could pose a potential challenge in the treatment of SLE, especially in terms of inducing B cell tolerance. Additionally, it has been postulated that Th17 cells, a subset of T cells, might be responsible for the abnormal selection of autoreactive B cells in GCs and the humoral response in vivo [79][78]. Collectively, inhibiting the differentiation of Tfh cells into Th17 cells and B cells into long-lived plasma cells, alongside expanding Tregs, could emerge as novel treatment options for SLE, centered on diminishing autoantibody production. Furthermore, the expansion of Treg cells through low-dose IL-2 has been shown to alleviate hypertension in NZBWF1 mice [80][79], while the neutralization of IL-17, the primary proinflammatory cytokine produced by Th17 cells, has been found to ameliorate endothelial dysfunction and reduce high blood pressure in SLE mice induced by TLR7 activation [18][31].References
- Tsokos, G.C. Systemic Lupus Erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121.
- Yu, C.; Gershwin, M.E.; Chang, C. Diagnostic criteria for systemic lupus erythematosus: A critical review. J. Autoimmun. 2014, 48–49, 10–13.
- Akhil, A.; Bansal, R.; Anupam, K.; Tandon, A.; Bhatnagar, A. Systemic lupus erythematosus: Latest insight into etiopathogenesis. Rheumatol. Int. 2023, 43, 1381–1393.
- Wang, H.; Li, T.; Chen, S.; Gu, Y.; Ye, S. Neutrophil Extracellular Trap Mitochondrial DNA and Its Autoantibody in Systemic Lupus Erythematosus and a Proof-of-Concept Trial of Metformin. Arthritis Rheumatol. 2015, 67, 3190–3200.
- Blachut, D.; Przywara-Chowaniec, B.; Tomasik, A.; Kukulski, T.; Morawiec, B. Update of Potential Biomarkers in Risk Prediction and Monitoring of Atherosclerosis in Systemic Lupus Erythematosus to Prevent Cardiovascular Disease. Biomedicines 2023, 11, 2814.
- Tselios, K.; Koumaras, C.; Gladman, D.D.; Urowitz, M.B. Dyslipidemia in systemic lupus erythematosus: Just another comorbidity? Semin. Arthritis Rheum. 2016, 45, 604–610.
- Urowitz, M.B.; Gladman, D.; Ibañez, D.; Fortin, P.; Sanchez-Guerrero, J.; Bae, S.; Clarke, A.; Bernatsky, S.; Gordon, C.; Hanly, J.; et al. Accumulation of coronary artery disease risk factors over three years: Data from an international inception cohort. Arthritis Care Res. 2008, 59, 176–180.
- Urowitz, M.B.; Gladman, D.; Ibañez, D.; Fortin, P.; Sanchez-Guerrero, J.; Bae, S.; Clarke, A.; Bernatsky, S.; Gordon, C.; Hanly, J.; et al. Clinical manifestations and coronary artery disease risk factors at diagnosis of systemic lupus erythematosus: Data from an international inception cohort. Lupus 2007, 16, 731–735.
- Mok, C.C. Metabolic syndrome and systemic lupus erythematosus: The connection. Expert. Rev. Clin. Immunol. 2019, 15, 765–775.
- Bello, N.; Meyers, K.J.; Workman, J.; Hartley, L.; McMahon, M. Cardiovascular events and risk in patients with systemic lupus erythematosus: Systematic literature review and meta-analysis. Lupus 2023, 32, 325–341.
- Esdaile, J.M.; Abrahamowicz, M.; Grodzicky, T.; Li, Y.; Panaritis, C.; Berger, R.D.; Côte, R.; Grover, S.A.; Fortin, P.R.; Clarke, A.E.; et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum. 2001, 44, 2331–2337.
- Frostegård, J. Systemic lupus erythematosus and cardiovascular disease. J. Intern. Med. 2023, 293, 48–62.
- Zhao, M.; Feng, R.; Werth, V.P.; Williams, K.J. State of current management of the heightened risk for atherosclerotic cardiovascular events in an established cohort of patients with lupus erythematosus. Lupus Sci. Med. 2023, 10, e000908.
- Tselios, K.; Koumaras, C.; Urowitz, M.B.; Gladman, D.D. Do current arterial hypertension treatment guidelines apply to systemic lupus erythematosus patients? A critical appraisal. Semin. Arthritis Rheum. 2014, 43, 521–525.
- Ikdahl, E.; Wibetoe, G.; Rollefstad, S.; Salberg, A.; Bergsmark, K.; Kvien, T.K.; Olsen, I.C.; Soldal, D.M.; Bakland, G.; Lexberg, Å.; et al. Guideline recommended treatment to targets of cardiovascular risk is inadequate in patients with inflammatory joint diseases. Int. J. Cardiol. 2019, 274, 311–318.
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788.
- Manzi, S.; Meilahn, E.N.; Rairie, J.E.; Conte, C.G.; Medsger, T.A.; Jansen-McWilliams, L.; D’Agostino, R.B.; Kuller, L.H. Age-specific Incidence Rates of Myocardial Infarction and Angina in Women with Systemic Lupus Erythematosus: Comparison with the Framingham Study. Am. J. Epidemiol. 1997, 145, 408–415.
- Taylor, E.B.; Ryan, M.J. Understanding mechanisms of hypertension in systemic lupus erythematosus. Ther. Adv. Cardiovasc. Dis. 2017, 11, 20–32.
- Small, H.Y.; Migliarino, S.; Czesnikiewicz-Guzik, M.; Guzik, T.J. Hypertension: Focus on autoimmunity and oxidative stress. Free Radic. Biol. Med. 2018, 125, 104–115.
- Gonzalez-Vicente, A.; Hong, N.; Garvin, J.L. Effects of reactive oxygen species on renal tubular transport. Am. J. Physiol. Ren. Physiol. 2019, 317, F444–F455.
- Burnett, R.; Ravel, G.; Descotes, J. Clinical and histopathological progression of lesions in lupus-prone (NZB×NZW) F1 mice. Exp. Toxicol. Pathol. 2004, 56, 37–44.
- Chaudhari, S.; Pham, G.S.; Brooks, C.D.; Dinh, V.Q.; Young-Stubbs, C.M.; Shimoura, C.G.; Mathis, K.W. Should Renal Inflammation Be Targeted While Treating Hypertension? Front. Physiol. 2022, 13, 886779.
- Richard, M.L.; Gilkeson, G. Mouse models of lupus: What they tell us and what they don’t. Lupus Sci. Med. 2018, 5, e000199.
- Mathis, K.W.; Venegas-Pont, M.; Masterson, C.W.; Wasson, K.L.; Ryan, M.J. Blood pressure in a hypertensive mouse model of SLE is not salt-sensitive. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2011, 301, R1281–R1285.
- Herlitz, H.; Svalander, C.; Tarkowski, A.; Westberg, G. Effect of captopril on murine systemic lupus erythematosus disease. J. Hypertens. 1988, 6, S684–S686.
- Herlitz, H.; Tarkowski, A.; Svalander, C.; Volkmann, R.; Westberg, G. Beneficial Effect of Captopril on Systemic Lupus erythematosus-Like Disease in MRL 1pr/1pr Mice. Int. Arch. Allergy Immunol. 1988, 85, 272–277.
- De Albuquerque, D.A.; Saxena, V.; Adams, D.E.; Boivin, G.P.; Brunner, H.I.; Witte, D.P.; Singh, R.R. An ACE inhibitor reduces Th2 cytokines and TGF-β1 and TGF-β2 isoforms in murine lupus nephritis. Kidney Int. 2004, 65, 846–859.
- Yokogawa, M.; Takaishi, M.; Nakajima, K.; Kamijima, R.; Fujimoto, C.; Kataoka, S.; Terada, Y.; Sano, S. Epicutaneous Application of Toll-like Receptor 7 Agonists Leads to Systemic Autoimmunity in Wild-Type Mice: A New Model of Systemic Lupus Erythematosus. Arthritis Rheumatol. 2014, 66, 694–706.
- Villalvazo, P.; Carriazo, S.; Rojas-Rivera, J.; Ramos, A.M.; Ortiz, A.; Perez-Gomez, M.V. Gain-of-function TLR7 and loss-of-function A20 gene variants identify a novel pathway for Mendelian lupus and lupus nephritis. Clin. Kidney J. 2022, 15, 1973–1980.
- Liu, Y.; Seto, N.L.; Carmona-Rivera, C.; Kaplan, M.J. Accelerated model of lupus autoimmunity and vasculopathy driven by toll-like receptor 7/9 imbalance. Lupus Sci. Med. 2018, 5, e000259.
- Robles-Vera, I.; La Visitación, N.D.; Toral, M.; Sánchez, M.; Gómez-Guzmán, M.; O’valle, F.; Jiménez, R.; Duarte, J.; Romero, M. Toll-like receptor 7-driven lupus autoimmunity induces hypertension and vascular alterations in mice. J. Hypertens. 2020, 38, 1322–1335.
- Sandling, J.K.; Pucholt, P.; Rosenberg, L.H.; Farias, F.H.G.; Kozyrev, S.V.; Eloranta, M.-L.; Alexsson, A.; Bianchi, M.; Padyukov, L.; Bengtsson, C.; et al. Molecular pathways in patients with systemic lupus erythematosus revealed by gene-centred DNA sequencing. Ann. Rheum. Dis. 2021, 80, 109–117.
- McClung, D.M.; Kalusche, W.J.; Jones, K.E.; Ryan, M.J.; Taylor, E.B. Hypertension and endothelial dysfunction in the pristane model of systemic lupus erythematosus. Physiol. Rep. 2021, 9, e14734.
- Manzi, S.; Selzer, F.; Sutton-Tyrrell, K.; Fitzgerald, S.G.; Rairie, J.E.; Tracy, R.P.; Kuller, L.H. Prevalence and risk factors of carotid plaque in women with systemic lupus erythematosus. Arthritis Rheum. 1999, 42, 51–60.
- Clemmer, J.S.; Hillegass, W.B.; Taylor, E.B. Antihypertensive effects of immunosuppressive therapy in autoimmune disease. J. Hum. Hypertens. 2022, 37, 300–306.
- Gómez-Guzmán, M.; Jiménez, R.; Romero, M.; Sánchez, M.; Zarzuelo, M.J.; Gómez-Morales, M.; O’Valle, F.; López-Farré, A.J.; Algieri, F.; Gálvez, J.; et al. Chronic hydroxychloroquine improves endothelial dysfunction and protects kidney in a mouse model of systemic lupus erythematosus. Hypertension 2014, 64, 330–337.
- Virdis, A.; Tani, C.; Duranti, E.; Vagnani, S.; Carli, L.; Kühl, A.A.; Solini, A.; Baldini, C.; Talarico, R.; Bombardieri, S.; et al. Early treatment with hydroxychloroquine prevents the development of endothelial dysfunction in a murine model of systemic lupus erythematosus. Arthritis Res. Ther. 2015, 17, 277.
- Miranda, S.; Billoir, P.; Damian, L.; Thiebaut, P.A.; Schapman, D.; Le Besnerais, M.; Jouen, F.; Galas, L.; Levesque, H.; Le Cam-Duchez, V.; et al. Hydroxychloroquine reverses the prothrombotic state in a mouse model of antiphospholipid syndrome: Role of reduced inflammation and endothelial dysfunction. PLoS ONE 2019, 14, e0212614.
- Furumoto, Y.; Smith, C.K.; Blanco, L.; Zhao, W.; Brooks, S.R.; Thacker, S.G.; Zarzour, A.; Sciumè, G.; Tsai, W.L.; Trier, A.M.; et al. Tofacitinib Ameliorates Murine Lupus and Its Associated Vascular Dysfunction. Arthritis Rheumatol. 2017, 69, 148–160.
- Hasni, S.A.; Gupta, S.; Davis, M.; Poncio, E.; Temesgen-Oyelakin, Y.; Carlucci, P.M.; Wang, X.; Naqi, M.; Playford, M.P.; Goel, R.R.; et al. Phase 1 double-blind randomized safety trial of the Janus kinase inhibitor tofacitinib in systemic lupus erythematosus. Nat. Commun. 2021, 12, 3391.
- Verden, A.; Dimbil, M.; Kyle, R.; Overstreet, B.; Hoffman, K.B. Analysis of Spontaneous Postmarket Case Reports Submitted to the FDA Regarding Thromboembolic Adverse Events and JAK Inhibitors. Drug Saf. 2018, 41, 357–361.
- Rajagopalan, S.; Somers, E.C.; Brook, R.D.; Kehrer, C.; Pfenninger, D.; Lewis, E.; Chakrabarti, A.; Richardson, B.C.; Shelden, E.; McCune, W.J.; et al. Endothelial cell apoptosis in systemic lupus erythematosus: A common pathway for abnormal vascular function and thrombosis propensity. Blood 2004, 103, 3677–3683.
- Nikpour, M.; Gladman, D.D.; Ibañez, D.; Bruce, I.N.; Burns, R.J.; Urowitz, M.B. Myocardial Perfusion Imaging in Assessing Risk of Coronary Events in Patients with Systemic Lupus Erythematosus. J. Rheumatol. 2009, 36, 288–294.
- Mak, A.; Kow, N.Y.; Schwarz, H.; Gong, L.; Tay, S.H.; Ling, L.H. Endothelial dysfunction in systemic lupus erythematosus—A case-control study and an updated meta-analysis and meta-regression. Sci. Rep. 2017, 7, 7320.
- Ryan, M.J.; McLemore, G.R. Hypertension and impaired vascular function in a female mouse model of systemic lupus erythematosus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R736–R742.
- Romero, M.; Toral, M.; Robles-Vera, I.; Sánchez, M.; Jiménez, R.; O’Valle, F.; Rodriguez-Nogales, A.; Pérez-Vizcaino, F.; Gálvez, J.; Duarte, J. Activation of Peroxisome Proliferator Activator Receptor β/δ Improves Endothelial Dysfunction and Protects Kidney in Murine Lupus. Hypertension 2017, 69, 641–650.
- Montoya, T.; Sánchez-Hidalgo, M.; Castejón, M.L.; Vazquéz-Román, M.V.; de Sotomayor, M.A.; Ortega-Vidal, J.; González, M.L.; Alarcón-de-la-Lastra, C. Oleocanthal supplemented diet improves renal damage and endothelial dysfunction in pristane-induced systemic lupus erythematosus in mice. Food Res. Int. 2023, 163, 112140.
- Moleón, J.; González-Correa, C.; Robles-Vera, I.; Miñano, S.; de la Visitación, N.; Barranco, A.M.; Martín-Morales, N.; O’Valle, F.; Mayo-Martínez, L.; García, A.; et al. Targeting the gut microbiota with dietary fibers: A novel approach to prevent the development cardiovascular complications linked to systemic lupus erythematosus in a preclinical study. Gut Microbes 2023, 15, 2247053.
- Nguyen, H.; Chiasson, V.L.; Chatterjee, P.; Kopriva, S.E.; Young, K.J.; Mitchell, B.M. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc. Res. 2013, 97, 696–704.
- Pietrowski, E.; Bender, B.; Huppert, J.; White, R.; Luhmann, H.J.; Kuhlmann, C.R.W. Pro-Inflammatory Effects of Interleukin-17A on Vascular Smooth Muscle Cells Involve NAD(P)H- Oxidase Derived Reactive Oxygen Species. J. Vasc. Res. 2011, 48, 52–58.
- Kassan, M.; Galan, M.; Partyka, M.; Trebak, M.; Matrougui, K. Interleukin-10 Released by CD4 + CD25 + Natural Regulatory T Cells Improves Microvascular Endothelial Function Through Inhibition of NADPH Oxidase Activity in Hypertensive Mice. Arter. Thromb. Vasc. Biol. 2011, 31, 2534–2542.
- Liang, C.-F.; Liu, J.T.; Wang, Y.; Xu, A.; Vanhoutte, P.M. Toll-Like Receptor 4 Mutation Protects Obese Mice Against Endothelial Dysfunction by Decreasing NADPH Oxidase Isoforms 1 and 4. Arter. Thromb. Vasc. Biol. 2013, 33, 777–784.
- Toral, M.; Jiménez, R.; Romero, M.; Robles-Vera, I.; Sánchez, M.; Salaices, M.; Sabio, J.M.; Duarte, J. Role of endoplasmic reticulum stress in the protective effects of PPARβ/δ activation on endothelial dysfunction induced by plasma from patients with lupus. Arthritis Res. Ther. 2017, 19, 268.
- Pieterse, E.; Rother, N.; Garsen, M.; Hofstra, J.M.; Satchell, S.C.; Hoffmann, M.; Loeven, M.A.; Knaapen, H.K.; van der Heijden, O.W.H.; Berden, J.H.M.; et al. Neutrophil Extracellular Traps Drive Endothelial-to-Mesenchymal Transition. Arter. Thromb. Vasc. Biol. 2017, 37, 1371–1379.
- Lee, P.Y.; Li, Y.; Richards, H.B.; Chan, F.S.; Zhuang, H.; Narain, S.; Butfiloski, E.J.; Sobel, E.S.; Reeves, W.H.; Segal, M.S. Type I interferon as a novel risk factor for endothelial progenitor cell depletion and endothelial dysfunction in systemic lupus erythematosus. Arthritis Rheum. 2007, 56, 3759–3769.
- Thacker, S.; Duquaine, D.; Park, J.; Kaplan, M. Lupus-prone New Zealand Black/New Zealand White F1 mice display endothelial dysfunction and abnormal phenotype and function of endothelial progenitor cells. Lupus 2010, 19, 288–299.
- Geng, L.; Wang, S.; Li, X.; Wang, D.; Chen, H.; Chen, J.; Sun, Y.; Chen, W.; Yao, G.; Gao, X.; et al. Association between Type I interferon and depletion and dysfunction of endothelial progenitor cells in C57BL/6 mice deficient in both apolipoprotein E and Fas ligand. Curr. Res. Transl. Med. 2018, 66, 71–82.
- Buie, J.J.; Renaud, L.L.; Muise-Helmericks, R.; Oates, J.C. IFN-α Negatively Regulates the Expression of Endothelial Nitric Oxide Synthase and Nitric Oxide Production: Implications for Systemic Lupus Erythematosus. J. Immunol. 2017, 199, 1979–1988.
- Sacre, K.; Escoubet, B.; Pasquet, B.; Chauveheid, M.-P.; Zennaro, M.-C.; Tubach, F.; Papo, T. Increased Arterial Stiffness in Systemic Lupus Erythematosus (SLE) Patients at Low Risk for Cardiovascular Disease: A Cross-Sectional Controlled Study. PLoS ONE 2014, 9, e94511.
- Shang, Q.; Tam, L.; Li, E.; Yip, G.; Yu, C. Increased arterial stiffness correlated with disease activity in systemic lupus erythematosus. Lupus 2008, 17, 1096–1102.
- Diószegi, Á.; Lőrincz, H.; Kaáli, E.; Soltész, P.; Perge, B.; Varga, É.; Harangi, M.; Tarr, T. Role of Altered Metabolism of Triglyceride-Rich Lipoprotein Particles in the Development of Vascular Dysfunction in Systemic Lupus Erythematosus. Biomolecules 2023, 13, 401.
- Rodriguez-Iturbe, B.; Pons, H.; Johnson, R.J. Role of the Immune System in Hypertension. Physiol. Rev. 2017, 97, 1127–1164.
- Khraibi, A.A.; Norman, R.A.; Dzielak, D.J. Chronic immunosuppression attenuates hypertension in Okamoto spontaneously hypertensive rats. Am. J. Physiol.-Heart Circ. Physiol. 1984, 247, H722–H726.
- Robles-Vera, I.; de la Visitación, N.; Sánchez, M.; Gómez-Guzmán, M.; Jiménez, R.; Moleón, J.; González-Correa, C.; Romero, M.; Yang, T.; Raizada, M.K.; et al. Mycophenolate Improves Brain–Gut Axis Inducing Remodeling of Gut Microbiota in DOCA-Salt Hypertensive Rats. Antioxidants 2020, 9, 1199.
- Robles-Vera, I.; de la Visitación, N.; Toral, M.; Sánchez, M.; Gómez-Guzmán, M.; Jiménez, R.; Romero, M.; Duarte, J. Mycophenolate mediated remodeling of gut microbiota and improvement of gut-brain axis in spontaneously hypertensive rats. Biomed. Pharmacother. 2021, 135, 111189.
- Tipton, A.J.; Baban, B.; Sullivan, J.C. Female spontaneously hypertensive rats have greater renal anti-inflammatory T lymphocyte infiltration than males, American Journal of Physiology-Regulatory. Integr. Comp. Physiol. 2012, 303, R359–R367.
- Mathis, K.W.; Wallace, K.; Flynn, E.R.; Maric-Bilkan, C.; LaMarca, B.; Ryan, M.J. Preventing Autoimmunity Protects Against the Development of Hypertension and Renal Injury. Hypertension 2014, 64, 792–800.
- Taylor, E.B.; Ryan, M.J. Immunosuppression With Mycophenolate Mofetil Attenuates Hypertension in an Experimental Model of Autoimmune Disease. J. Am. Heart Assoc. 2017, 6, e005394.
- Kamal, A.; Khamashta, M. The efficacy of novel B cell biologics as the future of SLE treatment: A review. Autoimmun. Rev. 2014, 13, 1094–1101.
- Taylor, E.B.; Barati, M.T.; Powell, D.W.; Turbeville, H.R.; Ryan, M.J. Plasma Cell Depletion Attenuates Hypertension in an Experimental Model of Autoimmune Disease. Hypertension 2018, 71, 719–728.
- Liu, Z.; Bethunaickan, R.; Huang, W.; Lodhi, U.; Solano, I.; Madaio, M.P.; Davidson, A. Interferon-α accelerates murine systemic lupus erythematosus in a T cell-dependent manner. Arthritis Rheum. 2011, 63, 219–229.
- Jacob, J.; Kelsoe, G.; Rajewsky, K.; Weiss, U. Intraclonal generation of antibody mutants in germinal centres. Nature 1991, 354, 389–392.
- MacLennan, I.C.M.; Toellner, K.-M.; Cunningham, A.F.; Serre, K.; Sze, D.M.-Y.; Zúñiga, E.; Cook, M.C.; Vinuesa, C.G. Extrafollicular antibody responses. Immunol. Rev. 2003, 194, 8–18.
- Vinuesa, C.G.; Cook, M.C.; Angelucci, C.; Athanasopoulos, V.; Rui, L.; Hill, K.M.; Yu, D.; Domaschenz, H.; Whittle, B.; Lambe, T.; et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature 2005, 435, 452–458.
- Subramanian, S.; Woolford, C.A.; Drill, E.; Lu, M.; Jones, E.W. Pbn1p: An essential endoplasmic reticulum membrane protein required for protein processing in the endoplasmic reticulum of budding yeast. Proc. Natl. Acad. Sci. USA 2006, 103, 939–944.
- Seo, S.; Fields, M.L.; Buckler, J.L.; Reed, A.J.; Mandik-Nayak, L.; Nish, S.A.; Noelle, R.J.; Turka, L.A.; Finkelman, F.D.; Caton, A.J.; et al. The Impact of T Helper and T Regulatory Cells on the Regulation of Anti-Double-Stranded DNA B Cells. Immunity 2002, 16, 535–546.
- Lee, H.-Y.; Hong, Y.-K.; Yun, H.-J.; Kim, Y.-M.; Kim, J.-R.; Yoo, W.-H. Altered frequency and migration capacity of CD4+CD25+ regulatory T cells in systemic lupus erythematosus. Rheumatology 2008, 47, 789–794.
- Hsu, H.-C.; Yang, P.; Wang, J.; Wu, Q.; Myers, R.; Chen, J.; Yi, J.; Guentert, T.; Tousson, A.; Stanus, A.L.; et al. Interleukin 17–producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat. Immunol. 2008, 9, 166–175.
- Taylor, E.B.; Sasser, J.M.; Maeda, K.J.; Ryan, M.J. Expansion of regulatory T cells using low-dose interleukin-2 attenuates hypertension in an experimental model of systemic lupus erythematosus. Am. J. Physiol.-Ren. Physiol. 2019, 317, F1274–F1284.
More