Pulmonary hypertension (PH) is a disorder involving a heterogeneous group of medical conditions encompassing several cardiopulmonary illnesses.
1. Introduction
Pulmonary hypertension (PH) is a disorder involving a heterogeneous group of medical conditions encompassing several cardiopulmonary illnesses. The most recent 6th World Symposium on Pulmonary Hypertension redefined the threshold for recognizing PH, including a new cut-off level for mean pulmonary artery pressure ≥20 mmHg
[1]. The World Health Organization’s diagnostic groups of PH provide a useful framework for categorizing the various etiologies of PH, whereas the hemodynamics more directly allow us to understand the phenotype (i.e., precapillary, postcapillary, or combined pre- and postcapillary).
The diagnosis of PH can be complex, and at times, it requires a multidisciplinary approach in order to detect and manage PH. General practitioners are frequently the first physicians to encounter this group of patients
[2]. Despite the life-threatening nature of this condition and the increased awareness and advances in therapies in the past 20 years, significant delays from the onset of symptoms to the time of diagnosis remain. The time from symptom onset to a diagnosis of PH can be delayed by a mean of over 2 years and occur after multiple hospitalizations. The highest likelihood of delayed recognition occurs in patients that are less than 36 years old. Deano et al.
[3] demonstrated that 60% of patients had functional class III or IV symptoms, and 33% were misdiagnosed at the time of referral for PH. These delays can result in a worsening clinical outcome or survival
[2][3][4][5][6][2,3,4,5,6].
2. Classification of PH
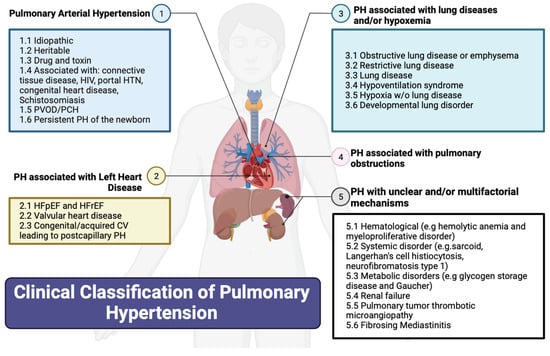
Figure 1 summarizes the updated classification of the 6th World Symposium on PH based on etiology. The WHO’s Group I PH, referred to as pulmonary arterial hypertension (PAH), encompasses various causes including connective tissue diseases (most commonly systemic sclerosis), HIV, portal hypertension, drug and toxin exposures, congenital heart diseases causing systemic-to-pulmonary shunting, and idiopathic and hereditary PH. PAH in particular is an underdiagnosed but serious disease, characterized by progressive right heart failure
[7][8]. The WHO’s Group II PH is PH from left heart diseases, also known as postcapillary or pulmonary venous hypertension and includes left ventricular systolic and/or diastolic heart failure and heart failure related to left-sided valvular disease. The WHO’s Group III PH is caused by chronic hypoxic and respiratory diseases. The WHO’s Group IV PH is related to chronic pulmonary artery obstructions, most commonly chronic thromboembolic pulmonary hypertension (CTEPH). Lastly, the WHO’s Group V PH includes miscellaneous diseases such as sarcoidosis, thyroid disorders, and end-stage renal disease with or without dialysis.
Figure 1. Clinical classification of pulmonary hypertension. CV: cardiovascular; e.g.: example given; HFpEF: heart failure with preserved ejection fraction; HFrEF: heart failure with reduced ejection fraction; HIV: human immunodeficiency virus; HTN: hypertension; PH: pulmonary hypertension; w/o: without. Created using Biorender.
3. History and Physical Exam
By far the most common and one of the earliest presenting symptoms is dyspnea with exertion. This can frequently be the only presenting symptom. The nonspecific nature of this symptom frequently results in misdiagnosis for more common disorders such as asthma, left heart failure, or deconditioning associated with obesity. Orthopnea is more commonly a feature of PH that is secondary to left heart disease as opposed to PAH. Exertional presyncope and syncope are hallmark symptoms of PAH and are frequently what draws attention to the diagnosis. Exertional syncope, as well as rapidly worsening functional capacity, are considered high-risk findings that warrant urgent intervention. Exertional chest pain, which is typically related to right ventricular (RV) ischemia related to limited coronary perfusion in the context of chamber enlargement and hypertrophy, is another common symptom that should be recognized as a manifestation of PAH. Historically, hoarseness (due to compression of the left laryngeal recurrent nerve) and wheezing (due to compression of the bronchi) have been described, but practically, they do not occur, and these symptoms should not be expected or associated with PAH
[1][8][9][1,9,10].
During a physical exam, there are multiple findings that can be elicited on cardiac auscultation. Though nonspecific for PH, the systolic murmur of tricuspid regurgitation may be auscultated. An increased pulmonic component to the second heart sound, related to the early closure of the pulmonic valve, may be appreciated. In the setting of RV hypertrophy (RVH) and enlargement, palpation over the sternum may reveal a prominent pulsation, termed the parasternal heave
[10][11]. As the PH syndrome advances, clinical findings of heart failure such as elevated jugular venous pressure, lower extremity edema, and ascites may be apparent. The jugular venous pulsation may have “V” waves, suggesting significant tricuspid regurgitation. The presence of significant right heart failure warrants urgent attention. Resting tachycardia, hypotension, and exertional hypoxia are signs of impaired cardiac output and pulmonary vascular disease and warrant urgent intervention
[1][10][1,11].
Elements from the patient’s history and physical exam may provide clues to the etiology. A history of Raynaud’s syndrome, dysphagia, or gastroesophageal reflux and physical findings of sclerodactyly or telangiectasias may suggest undiagnosed connective tissue disease. Digital clubbing may suggest a systemic-to-pulmonary shunt with Eisenmenger’s syndrome, which is associated with congenital heart disease or advanced lung disease. An extensive alcohol abuse history or methamphetamine use may suggest Group I PH, related to portopulmonary hypertension or toxins, respectively. Historical elements which predispose patients to left heart disease include traditional cardiovascular risk factors such as the presence of atherosclerosis, systemic arterial hypertension requiring two or more medications, atrial fibrillation, and obesity
[11][12]. An extensive smoking history, abnormal lung sounds, and profound hypoxia may suggest PH in the setting of chronic lung disease. A prior history of hypercoagulable disorders or history of venous thromboembolism may increase suspicion for CTEPH, but the absence of these does not rule out the likelihood of this diagnosis
[1][10][12][1,11,13].
4. Risk Factors for Pulmonary Hypertension
Certain conditions will be considered risk factors for PH, such as a prior history of pulmonary embolism, use of methamphetamine, connective tissue disease, portal hypertension, HIV, sarcoidosis, congenital heart disease, or a family history of PAH. Screening at-risk patients has been described. In the case of connective-tissue-disease-associated PAH, around 75% have systemic sclerosis, and it carries poor prognosis. Hence, the inclusion of the DETECT algorithm
[13][14] is a Class 1 recommendation in asymptomatic adults with systemic sclerosis of more than 3 years, FVC ≥ 40% and DLCO < 60%.