Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Lindsay Dong and Version 1 by Sergey Sedykh.
Plant growth-promoting bacteria are commonly used in agriculture, particularly for seed inoculation. Multispecies consortia are believed to be the most promising form of these bacteria. However, designing and modeling bacterial consortia to achieve desired phenotypic outcomes in plants is challenging.
- rhizobacteria
- microbe–plant interaction
- PGPB
- microbial communities
1. Introduction
Microbial communities associated with plants are called the plant microbiome [1]. They are critical for plant health and adaptation to environmental factors [2]. The interactions between plants and soil microbial communities can be very complex. When the soil microbiome is formed, plants select the microbial partners that could improve their growth, development, and productivity. The soil microbiota is supplied with nutrients secreted by plants as root exudates [3].
To date, only a few individual effects mutually exerted by plants and microbes have been well characterized, for example, nitrogen fixation by rhizobia. Developing novel strategies for more productive and sustainable agriculture requires understanding plant–microbe interactions and the possibility of modulating the plant microbiome [4,5][4][5]. Plant growth-promoting bacteria (PGPB) have been extensively studied.
Currently, most bacterial inoculants used to enhance crop productivity are based on a single strain with a set of plant growth-promoting traits [4,6,7][4][6][7]. Numerous characteristics of PGPB have been identified using in vitro screening assays or inoculation experiments under controlled conditions [8]. However, these characteristics are rarely tested in field conditions and related testing generally neglects the significant aspects of plant–microbe interactions [7].
Another strategy for developing microbial inoculants is to study microbial communities. The information about individual species and possible antagonistic interactions can be used to design synthetic microbial communities to be eventually modified into microbial communities with predictable traits. A key question is how to design bacterial consortia to produce the desired phenotypic outcomes for plants [9]. Not only is it necessary to understand how microbial communities interact and shape the plant microbiome. Moreover, it is of utmost importance to investigate the functions and potential contributions of individual microorganisms at the organismal and molecular levels. This will facilitate the development of manageable and traceable consortia with all the required properties for promoting plant growth [10].
Bacteria closely related to the plant rhizosphere are assumed to have a higher potential for interaction and are likely to contribute significantly to the host phenotype. Bacterial communities from the ecological niche of the rhizosphere have probably co-evolved with plants for a long time and share characteristic features such as metabolism, biofilm formation, and others not typical of individuals from other habitats, such as soil, sediments, marine ecosystems, and others [10].
2. Microbial Interactions
There are various forms of intermicrobial interactions, such as competition for resources, synergism, and antagonism, to name but a few. These interactions can be altered by the environment [57][11]. Soil bacteria are capable of distinguishing their microbial competitors and fine-tuning their survival strategies [58][12]. The secretion of some metabolites by soil microbes has been demonstrated to result directly from interactions with other microorganisms that are in close vicinity [59,60][13][14]. To illustrate, it has been discovered that soil bacteria, which do not seem to produce antimicrobials, begin synthesizing specific or broad-spectrum antibiotics when encountering other bacterial species in carbon-limited environments [61][15]. These findings emphasize the requirement for a comprehensive approach that could surpass the mere study of microbial activity. It is necessary to take into account or mimic the biotic and abiotic conditions of the soil niches commonly inhabited by microbes. A hypothesis exists that some microorganisms, referred to as beneficiaries, can “avoid” performing functions in order to optimize their adaptation to the environment. This function loss can be attributed to the presence of “helper” microorganisms performing the same function in the immediate vicinity, providing a stable environment. Hence, soil bacteria may exhibit a metabolic dependence on neighboring microbes. Although it is unknown whether this pattern is typical, it may explain why most soil microbes cannot be cultured individually under laboratory conditions [62][16]. It is probably for this reason that there is still a lack of physiological, morphological, and ecological characterization for many soil microorganisms due to the failure to isolate, culture, and study pure cultures. There are several strategies of interactions within natural consortia that enable the efficient utilization of resources. These interactions can be categorized following the classical ecological categories of interactions between organisms. Cooperation. Cooperative interactions are based on functional differentiation and specialization [63][17], contributing to an increase in overall resource efficiency and a decrease in the formation of byproducts. This type of interaction also allows for more optimal organization of several multi-step processes, such as the degradation of complex organic compounds. Cooperative interactions allocate carbon sources among community members in a non-competitive manner based on metabolic functionality. Cooperation allows parallel processing of the same substrate in different metabolic pathways and is used to create consortia that can simultaneously ferment pentose and hexose sugars, which is often unattainable in monocultures because of catabolic repression [64][18]. Commensalism. Another standard bacterial interaction mode is commensalism. This is when the activities of one community member provide an ecological niche for others with no benefit or cost to itself. One example of commensalism is metabolite exchange whereby a producer organism excretes byproducts with no benefit or cost to itself but provides metabolites to other members of the community [65][19]. Mutualism. Mutualism is a common natural phenomenon defined as a relationship that is beneficial to all participants. For example, a community member consuming organic acid removes inhibitory byproducts from a producer population [66][20]. Mutualistic interactions can be facultative or obligatory. In obligatory mutualistic interactions, organisms fail to perform a particular action when separated from the interaction partner. One example of such interactions is the syntrophy of secondary-fermenting bacteria with hydrogen-consuming microorganisms or methanotrophic Archaea with sulfate-reducing bacteria [67][21]. Competition. Competitive interactions, as well as predator–prey or parasite–prey interactions, are beneficial to only one interaction partner. Competition for nutrients is the most frequent antagonistic interaction in the microbial world [69][22]. In this case, microorganisms try to utilize nutrients while gaining advantages over other microorganisms. Microorganisms have developed several strategies for successful competition, such as biosynthesis and secretion of antibiotics, close association of enzymes of metabolic pathways, and uptake of degradation products. Parasitism. A parasitic interaction is established if the recipient forces the producer to release an intermediate compound to be used by the recipient. One example is the parasitic growth of Pseudomonas aeruginosa in co-culture with the chitinolytic bacterium Aeromonas hydrophila [70][23]. P. aeruginosa uses secondary metabolites to control metabolism in such a way that chitin is not completely oxidized to acetate, which serves as a substrate for Pseudomonas aeruginosa growth.3. Modeling Consortia
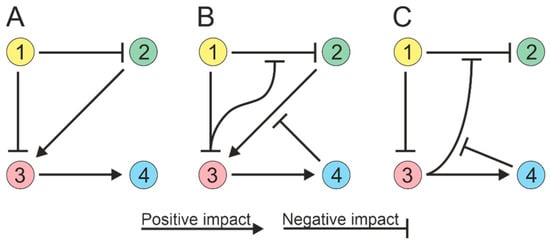
The process of modeling ecological interactions assumes pairwise interactions (Figure 21A). However, soil microorganisms can interact in higher-order combinations, i.e., interactions between two species can be regulated by other species [71,72][24][25]. For example, a microbial species can produce an antibiotic that inhibits the growth of competing species, and this inhibitory effect can be attenuated by a third species that produces an enzyme that cleaves the antibiotic [73,74][26][27]. Thus, the third species can alter the interactions between the antibiotic-producing strain and susceptible species without directly affecting either of them individually (Figure 21B). The activity of the enzyme responsible for antibiotic degradation can be impeded by compounds generated by the fourth species [75][28], resulting in a four-way interaction (Figure 21C). Thus, possible strategies for interspecific interactions must be considered when creating microbial communities.Figure 21. Soil microorganisms can exhibit high-order interactions, with interactions between two species being influenced by other species (1 to 4). (A) Paired interactions in a community; species directly influence each other. (B) Three-way community interactions; one species can modulate the interaction between two others. (C) Quadripartite community interactions.
Given the multitude of biochemical reactions conducted by the soil microbiome, researchers aim to establish model soil consortia to comprehensively investigate the metabolic interactions among species. Synthetic microbial communities are simplified ones. Ideally, a model consortium should comprise a manageable and reproducible number of species, allowing the population dynamics and specific metabolic and signaling interactions among its members to be experimentally analyzed [76,77][29][30]. The knowledge acquired from the analysis of these communities of reduced complexity will help to reveal details of metabolic pathways and interspecific interactions that occur in natural soil communities.
5.1. The “Bottom-Up” Approach
3.1. The “Bottom-Up” Approach
The “bottom-up” approach for analyzing the soil microbiome is to combine individual strains to form “synthetic” consortia [75,78][28][31]. There are a few drawbacks to be considered. It is crucial to know which species interact in the natural environment because there is a risk of combining species that do not interact in the natural habitat, which would lead to the obtainment of results that would be difficult to operationalize. Another disadvantage of this approach is the likelihood of establishing consortia with a limited number of members, indicating that only a fraction of the interacting soil components can be taken into account. Investigating synthetic communities has several advantages, and the simplicity of the analysis can be useful in unraveling particularly complex interactions. Creating a microbial system from one or more microorganisms allows one to construct a physical model with higher reproducibility, lower variability, and a lower number of unknowns than in the original system.
The first step is to narrow down the most important components of the microbial community and, as a consequence, their functions. As a result, one can identify and select the members of the microbial community that are crucial for the process of research interest, thereby significantly reducing the complexity of the system under study [79][32]. The second step is to analyze the ecologically relevant function of most soil microorganisms. Thus, soil microorganisms can be isolated, and those capable of performing different functions (e.g., nitrogen fixation, phosphate solubilization, siderophore synthesis, and auxin production) can be identified.
5.2. The “Top-Down” Approach
3.2. The “Top-Down” Approach
A “top-down” approach for investigating the diversity and complexity of the soil microbiome is to create low-complexity consortia [86][33]. This method utilizes dilution and cultivation on complex carbon sources (e.g., N-acetylglucosamine and chitin). Dilution promotes a reduction in species richness of the community, and a stabilization of the consortia (after about 3–5 weeks of incubation) yields several microbial communities. However, the resulting consortia are still comparatively complex and contain up to several hundred species [87][34]. By adopting such an approach, the community can independently shape the species composition of a consortium and enhance its growth through necessary interactions. It should be noted that the type of carbon source has an impact on the rate and direction of consortium development [88][35]. The “top-down” approach involves identifying the community function to understand the community structure and dynamics. The “top-down” approach outperforms a “bottom-up” approach in generating representative communities [90][36]. Moreover, the larger community allows more soil functional potential and species interactions to be captured and studied. For example, applying the “top-down” approach resulted in 35 species being detected, with approximately 13 being the dominant ones compared to 2–3 dominant ones detected using the “bottom-up” approach [87][34]. The “top-down” approach proves useful for studying relationships between key community properties, such as diversity, function, and stability, while also providing a mechanistic understanding of community structure [89][37]. It is not unlikely that difficult-to-cultivate species can be studied exclusively using this approach due to their need for interaction with other members and their inability to exist independently without additional biochemical information [76][29]. It is worth noting that employing the two approaches considered above has resulted in valuable discoveries in the plant microbiome [88][35]. Combining the strengths of both approaches to study one model community can be highly effective. The “top-down” approach can reduce the soil microbiome to a more manageable size and identify interacting members.5.3. Practical Applications of Synthetic/Artificial Microbial Communities
3.3. Practical Applications of Synthetic/Artificial Microbial Communities
Synthetic consortia are small consortia of microorganisms designed to mimic and analyze the microbiome function and structure in vivo. Such consortia represent a new frontier for synthetic biology as they allow more complex problems to be solved compared to monocultures. The purpose of creating synthetic consortia is to reduce the complexity of the natural microbial community while preserving some of the original interactions between microbes and hosts, providing a repertoire of functions that cannot be achieved by a single microorganism [91,92,93][38][39][40]. Creating synthetic microbial communities in the laboratory involves selecting certain microorganisms according to their ability to stimulate plant growth, protect plants from pathogen infestation, or provide nutrition to plants. It has been suggested that microbial communities with increased metabolic activity should be modeled by selecting species that are not functionally too close or too distant [95][41]. However, the universality of this hypothesis has yet to be confirmed experimentally. Strain selection is assumed to be based, among other things, on functional genes rather than on taxonomic classification [96][42]. Three consortia were developed based on paired and triple interactions of phosphate-solubilizing bacteria for wheat inoculation, with the first consortium comprising Enterobacter sp. and Ochrobactrum sp., the second consortium comprising Pantoea sp., Enterobacter sp., and Ochrobactrum sp., and the third consortium comprising Ochrobactrum sp., Pseudomonas sp., and Bacillus sp. Rhizosphere analysis revealed a significant increase in linear root growth after inoculation with these consortia A key factor in developing a microbial consortium, which is essential for the successful functioning of the included microorganisms, is the compatibility of the individual microorganisms. Bacteria of the genera Bacillus and Trichoderma were found to perform well both in consortia and in individual inoculation, and Pseudomonas was reported to perform better in synthetic consortia than when inoculated individually [104][43]. These properties encountered when creating consortia are likely to be the clue to utilizing PGPB for agroecosystems. The potential contribution of individual microorganisms can be assessed by altering the composition of a synthetic community through the addition, removal, or replacement of microorganisms [10].5.4. Identification of Microbes with Key Characteristics
3.4. Identification of Microbes with Key Characteristics
Selecting microbes with important properties for agricultural applications mainly involves in vitro screening and sampling of well-known taxa or activities favorable to plant growth and development, such as atmospheric nitrogen fixation, phosphorus solubilization, phytohormone, and siderophore production [26,27][44][45]. Unfortunately, inoculating plants with preparations of microorganisms obtained using these traditional approaches often fails to result in long-term stable interactions with plants under field conditions and subsequently in satisfactory results [108,109][46][47]. For a strain to be considered successful, it must effectively compete with existing microorganisms in the soil, establish a strong presence in the plant root system, and form stable and sustainable associations, even in the face of potential changes in the environment and soil microbial composition throughout the growing season. One strategy to overcome the challenges is to select microorganisms based on the diversity of plant microbiota. The analysis of 16S rRNA gene sequencing data has revealed that certain groups of soil bacteria can successfully colonize plant roots and establish and maintain permanent relationships with them, whatever the environmental changes or the plant developmental stages [110,111][48][49]. Incorporating members of dominant groups referred to as the “core microbiome” into synthetic communities can mitigate the potential inefficiencies observed when strains are outcompeted by the natural soil microbiota.5.5. Development and Stabilization of Microbial Communities
3.5. Development and Stabilization of Microbial Communities
Synthetic microbial communities can be stabilized in the short term, up to 36 h, and in the long term, up to two weeks [110,115][48][50]. Given these data, consortia need to be given enough time to stabilize, e.g., several weeks, especially when using a “top-down” approach, before they can be maintained and revived. Significant changes in the relative abundance of some taxa are observed during the first week of incubation in soil, followed by stabilization after two weeks [87][34]. Stabilizing the soil microorganism community may require 3 to 5 weeks, with stability observed for several months afterward [86][33]. Thus, when studying model consortia, it is worth considering the period required to achieve stability after inoculating the consortium into new soil.4. Designing PGPB Consortia That Can Reduce the Response to Abiotic Stress
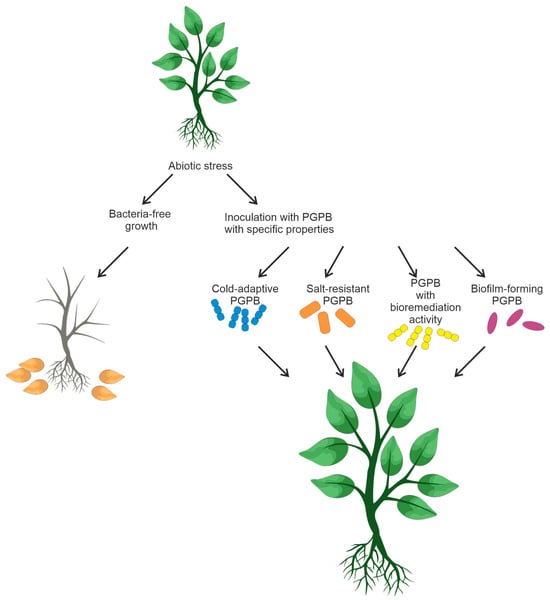
The design and creation of bacterial consortia is a non-trivial task that demands careful consideration. It is crucial to ensure the absence of antagonistic interactions among the members of the bacterial mixture to enable their coexistence. Moreover, bacteria selected for bacterial mixtures should be capable of enhancing plant growth, bioremediation, and tolerance of unfavorable conditions in crop fields. Figure 42 summarizes the information on soil bacteria and their possible consortia that could favorably affect plant growth.Figure 42.
Food crop productivity is affected by various abiotic factors, such as salinity, drought, and temperature [111,116][49][51]. Some functional features of rhizobacteria (biofilm formation, bioremediation, resistance to soil salinity, and low temperatures) have an impact on plant survival under unfavorable conditions [117,118][52][53]. PGPR-based microbial strategies can be efficient in overcoming the negative effects of abiotic factors. Some bacteria are capable of forming a biofilm that can enhance resistance to antibiotics, heat, UV radiation, and other environmental stresses [119,120][54][55]. When exposed to unfavorable conditions, soil bacteria secrete exopolysaccharides that form an organomineral envelope, also known as biofilm. These polysaccharides consist of complex mixtures of high-molecular-weight polymers (MW ≥ 10,000) [121][56].
It is common for bacteria to produce exopolysaccharides under conditions of heavy metal stress and high salinity [123][57]. However, high-quality exopolysaccharides can only be produced by halo- or drought-tolerant rhizobacteria that can tolerate and survive under harsh conditions [124][58].
Soil salinity is regarded as the most significant among abiotic factors [127][59]. Experiments on inoculation of salt tolerant PGPB have demonstrated remarkable results in increasing agricultural productivity of saline soils [128,129][60][61]. The mechanisms contributing to plant growth stimulation have already been reviewed [26,27][44][45]. Aside from the mechanisms inherent in PGPB, salt-resistant bacteria exhibit specialized mechanisms essential for salt stress tolerance and plant growth promotion
Temperature is another crucial factor affecting plant growth. Low temperature puts plants under stress and affects microbial growth and activity. Agricultural cultivation in cold regions requires biofertilizers that can work at low temperatures. Inoculation of cold-adapted microbial inoculants PGPB significantly promotes plant growth under low-temperature conditions [136][62]. Cold-adapted PGPR, such as Burkholderia and Pseudomonas spp., have been described [137,138][63][64].
Inoculation of plants with soil PGPB capable of reducing abiotic stress.
5. Multi-Omics Approaches for Studying Microorganisms and Their Consortia
Traditional microbial culturing methods and modern methods such as pyrosequencing or NGS [141,142][65][66] are used to characterize and compare microbial communities and identify individual functions in different ecological niches. These methods ha ve specific advantages and limitations that should be taken into account when designing consortia. It is essential to isolate pure cultures of bacteria in order to study them in detail, identify specific traits, and directly determine the genetic components that underlie useful phenotypes [143,144][67][68]. However, only some soil microorganisms can be cultured in laboratory conditions. Metagenomic analysis approaches and omics technologies allow the structure of the microbial community and the functions of its individual components to be determined [145,146][69][70]. “Collective phenotypes” of interacting species within the soil microbiome are referred to as the metaphenome [147][71]. Complete metagenomic sequencing provides information on the genomes of all microorganisms and allows the functional and metabolic potential of a community to be characterized [150][72]. However, not all genes are expressed at any given time, and the total DNA extracted from soil may contain sequences from currently inactive populations. Nevertheless, high-throughput approaches have identified functional signatures of some rhizosphere and endosphere communities [142,151][66][73], and it is this approach that can answer the question of which microbial genes are enriched in a particular microhabitat. The soil metatranscriptomics is used to determine the functions performed by individual members of the soil microbiome [152,153][74][75]. Metaproteogenomic approaches enable the exploration of active functions and metabolic pathways [154][76]. These two approaches provide insight into the timing and location of specific gene expression. However, such studies are usually based on relatively shallow metatranscriptomes with read depths insufficient to cover all the members of a community; thus, these studies investigate only the most abundant and transcriptionally active species and genes Metaproteomic measurements of microbial communities do provide reliable and reasonably accurate estimates of microbial population sizes [157][77] because proteins make up to 40–60% of bacterial cell biomass [158][78] and have a linear correlation with cell mass and volume [159][79]. Estimates of relative cell population sizes in the organism community, based on the summation of protein abundance or content, suggest that the habitat has achieved population equilibrium and stabilization. Metaproteomics allows large-scale identification and quantification of microbial community proteins, facilitating the characterization of microbial identity, functional roles, and interspecific interactions in the community [160][80].6. Analysis of Metabolic Networks of Microbial Communities
Reconstructing metabolic networks of microbial communities is a challenging task. Even when dealing with a single species, the iterative process of genome-wide reconstruction of the metabolic network demands a substantial amount of time [168][81]. Modeling a community is a more challenging process due to the increased complexity associated with interactions between species. The traditional practice of designing community metabolic networks focuses on reconstructing high-quality individual networks so that their combination may provide quantitative predictions of metabolic interactions and community behavior [169][82]. However, in practice, such an approach does not take into account possible microbial interactions. The minimum information required includes the genome sequence determining the key metabolic functions and physiological data, such as growth conditions for more accurate modeling of networks. Current sequencing methods fail to read the entire genome at a time. Therefore, all sequencing protocols start by cutting DNA into smaller fragments that the sequencer can read [172][83]. The sequences of overlapping fragments resulting from sequencing are called contigs. If the sequencing coverage is deep enough, contigs can be assembled into one or more scaffolds covering the complete genome. Subsequently, it is essential to ascertain the location of the genes and comprehend their respective functions. The most important genes for metabolic reconstruction are those that function as enzymes and transport proteins.7. Conclusions
Modern agricultural practices commonly make use of inoculants consisting of a single strain isolated through in vitro screening of plant growth stimulation activity or inoculation experiments under controlled conditions. Although these strategies are widely used, they neglect important aspects of plant–microbe interactions. Due to the highly diverse and complex nature of the plant rhizosphere microbiome, which is sustained through extensive interactions between microbes and their hosts, a more comprehensive understanding can only be achieved by implementing sophisticated research methodologies. In recent years, the studies of the plant rhizosphere microbiome have provided new insights into microbial diversity, abundance, distribution, dynamics, and functions. The emergence of various microbiome-related phenotypes is attributed not to the impact of a single species but to the cooperative interaction of multiple species that effectively execute a common function. Microbial interaction networks in soil are often analyzed to detect the co-occurrence among community members and to identify the key taxa. These taxa may be critical to microbial communities, and their removal can cause dramatic shifts in microbial community structure and function. Given the complexity and context-specific nature of ecological interactions among microorganisms, which involve both structural and random interactions, it is often difficult to discern the contributions of different members within a microbial consortium to a particular function or phenotype.References
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The Rhizosphere Microbiome: Significance of Plant Beneficial, Plant Pathogenic, and Human Pathogenic Microorganisms. FEMS Microbiol. Rev. 2013, 37, 634–663.
- Yukun, G.; Jianghui, C.; Genzeng, R.; Shilin, W.; Puyuan, Y.; Congpei, Y.; Hongkai, L.; Jinhua, C. Changes in the Root-Associated Bacteria of Sorghum Are Driven by the Combined Effects of Salt and Sorghum Development. Environ. Microbiome 2021, 16, 14.
- Zhang, J.; Cook, J.; Nearing, J.T.; Zhang, J.; Raudonis, R.; Glick, B.R.; Langille, M.G.I.; Cheng, Z. Harnessing the Plant Microbiome to Promote the Growth of Agricultural Crops. Microbiol. Res. 2021, 245, 126690.
- Finkel, O.M.; Castrillo, G.; Herrera Paredes, S.; Salas González, I.; Dangl, J.L. Understanding and Exploiting Plant Beneficial Microbes. Curr. Opin. Plant Biol. 2017, 38, 155–163.
- Benito, P.; Carro, L.; Bacigalupe, R.; Ortúzar, M.; Trujillo, M.E. From Roots to Leaves: The Capacity of Micromonospora to Colonize Different Legume Tissues. Phytobiomes J. 2022, 6, 35–44.
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; van Themaat, E.V.L.; Schulze-Lefert, P. Structure and Functions of the Bacterial Microbiota of Plants. Annu. Rev. Plant Biol. 2013, 64, 807–838.
- De Souza, R.S.C.; Armanhi, J.S.L.; Damasceno, N.D.B.; Imperial, J.; Arruda, P. Genome Sequences of a Plant Beneficial Synthetic Bacterial Community Reveal Genetic Features for Successful Plant Colonization. Front. Microbiol. 2019, 10, 1779.
- de Souza, R.S.C.; Armanhi, J.S.L.; Arruda, P. From Microbiome to Traits: Designing Synthetic Microbial Communities for Improved Crop Resiliency. Front. Plant Sci. 2020, 11, 1179.
- Herrera Paredes, S.; Gao, T.; Law, T.F.; Finkel, O.M.; Mucyn, T.; Teixeira, P.J.P.L.; Salas González, I.; Feltcher, M.E.; Powers, M.J.; Shank, E.A.; et al. Design of Synthetic Bacterial Communities for Predictable Plant Phenotypes. PLoS Biol. 2018, 16, e2003962.
- Levy, A.; Salas Gonzalez, I.; Mittelviefhaus, M.; Clingenpeel, S.; Herrera Paredes, S.; Miao, J.; Wang, K.; Devescovi, G.; Stillman, K.; Monteiro, F.; et al. Genomic Features of Bacterial Adaptation to Plants. Nat. Genet. 2018, 50, 138–150.
- Shrestha, H.K.; Appidi, M.R.; Villalobos Solis, M.I.; Wang, J.; Carper, D.L.; Burdick, L.; Pelletier, D.A.; Doktycz, M.J.; Hettich, R.L.; Abraham, P.E. Metaproteomics Reveals Insights into Microbial Structure, Interactions, and Dynamic Regulation in Defined Communities as They Respond to Environmental Disturbance. BMC Microbiol. 2021, 21, 308.
- Garbeva, P.; Silby, M.W.; Raaijmakers, J.M.; Levy, S.B.; de Boer, W. Transcriptional and Antagonistic Responses of Pseudomonas Fluorescens Pf0-1 to Phylogenetically Different Bacterial Competitors. ISME J. 2011, 5, 973–985.
- Traxler, M.F.; Watrous, J.D.; Alexandrov, T.; Dorrestein, P.C.; Kolter, R. Interspecies Interactions Stimulate Diversification of the Streptomyces Coelicolor Secreted Metabolome. MBio 2013, 4, e00459-13.
- Tyc, O.; van den Berg, M.; Gerards, S.; van Veen, J.A.; Raaijmakers, J.M.; de Boer, W.; Garbeva, P. Impact of Interspecific Interactions on Antimicrobial Activity among Soil Bacteria. Front. Microbiol. 2014, 5, 567.
- Garbeva, P.; de Boer, W. Inter-Specific Interactions between Carbon-Limited Soil Bacteria Affect Behavior and Gene Expression. Microb. Ecol. 2009, 58, 36–46.
- Tracanna, V.; Ossowicki, A.; Petrus, M.L.C.; Overduin, S.; Terlouw, B.R.; Lund, G.; Robinson, S.L.; Warris, S.; Schijlen, E.G.W.M.; van Wezel, G.P.; et al. Dissecting Disease-Suppressive Rhizosphere Microbiomes by Functional Amplicon Sequencing and 10× Metagenomics. mSystems 2021, 6, e01116-20.
- Briones, A.; Raskin, L. Diversity and Dynamics of Microbial Communities in Engineered Environments and Their Implications for Process Stability. Curr. Opin. Biotechnol. 2003, 14, 270–276.
- Eiteman, M.A.; Lee, S.A.; Altman, R.; Altman, E. A Substrate-Selective Co-Fermentation Strategy with Escherichia Coli Produces Lactate by Simultaneously Consuming Xylose and Glucose. Biotechnol. Bioeng. 2009, 102, 822–827.
- Ren, Z.; Ward, T.E.; Regan, J.M. Electricity Production from Cellulose in a Microbial Fuel Cell Using a Defined Binary Culture. Environ. Sci. Technol. 2007, 41, 4781–4786.
- Bernstein, H.C.; Paulson, S.D.; Carlson, R.P. Synthetic Escherichia Coli Consortia Engineered for Syntrophy Demonstrate Enhanced Biomass Productivity. J. Biotechnol. 2012, 157, 159–166.
- Milucka, J.; Ferdelman, T.G.; Polerecky, L.; Franzke, D.; Wegener, G.; Schmid, M.; Lieberwirth, I.; Wagner, M.; Widdel, F.; Kuypers, M.M.M. Zero-Valent Sulphur Is a Key Intermediate in Marine Methane Oxidation. Nature 2012, 491, 541–546.
- Ghoul, M.; Mitri, S. The Ecology and Evolution of Microbial Competition. Trends Microbiol. 2016, 24, 833–845.
- Jagmann, N.; Brachvogel, H.-P.; Philipp, B. Parasitic Growth of Pseudomonas Aeruginosa in Co-Culture with the Chitinolytic Bacterium Aeromonas Hydrophila. Environ. Microbiol. 2010, 12, 1787–1802.
- Poisot, T.; Stouffer, D.B.; Gravel, D. Beyond Species: Why Ecological Interaction Networks Vary through Space and Time. Oikos 2015, 124, 243–251.
- Wootton, J.T. Indirect Effects in Complex Ecosystems: Recent Progress and Future Challenges. J. Sea Res. 2002, 48, 157–172.
- Kelsic, E.D.; Zhao, J.; Vetsigian, K.; Kishony, R. Counteraction of Antibiotic Production and Degradation Stabilizes Microbial Communities. Nature 2015, 521, 516–519.
- Abrudan, M.I.; Smakman, F.; Grimbergen, A.J.; Westhoff, S.; Miller, E.L.; van Wezel, G.P.; Rozen, D.E. Socially Mediated Induction and Suppression of Antibiosis during Bacterial Coexistence. Proc. Natl. Acad. Sci. USA 2015, 112, 11054–11059.
- Lozano, G.L.; Bravo, J.I.; Garavito Diago, M.F.; Park, H.B.; Hurley, A.; Peterson, S.B.; Stabb, E.V.; Crawford, J.M.; Broderick, N.A.; Handelsman, J. Introducing THOR, a Model Microbiome for Genetic Dissection of Community Behavior. MBio 2019, 10, e02846-18.
- Zengler, K.; Hofmockel, K.; Baliga, N.S.; Behie, S.W.; Bernstein, H.C.; Brown, J.B.; Dinneny, J.R.; Floge, S.A.; Forry, S.P.; Hess, M.; et al. EcoFABs: Advancing Microbiome Science through Standardized Fabricated Ecosystems. Nat. Methods 2019, 16, 567–571.
- Kong, Z.; Hart, M.; Liu, H. Paving the Way From the Lab to the Field: Using Synthetic Microbial Consortia to Produce High-Quality Crops. Front. Plant Sci. 2018, 9, 1467.
- Rodríguez Amor, D.; Dal Bello, M. Bottom-Up Approaches to Synthetic Cooperation in Microbial Communities. Life 2019, 9, 22.
- Smercina, D.N.; Bailey, V.L.; Hofmockel, K.S. Micro on a Macroscale: Relating Microbial-Scale Soil Processes to Global Ecosystem Function. FEMS Microbiol. Ecol. 2021, 97, fiab091.
- Zegeye, E.K.; Brislawn, C.J.; Farris, Y.; Fansler, S.J.; Hofmockel, K.S.; Jansson, J.K.; Wright, A.T.; Graham, E.B.; Naylor, D.; McClure, R.S.; et al. Selection, Succession, and Stabilization of Soil Microbial Consortia. mSystems 2019, 4, e00055-19.
- McClure, R.; Naylor, D.; Farris, Y.; Davison, M.; Fansler, S.J.; Hofmockel, K.S.; Jansson, J.K. Development and Analysis of a Stable, Reduced Complexity Model Soil Microbiome. Front. Microbiol. 2020, 11, 1987.
- Großkopf, T.; Soyer, O.S. Synthetic Microbial Communities. Curr. Opin. Microbiol. 2014, 18, 72–77.
- Tecon, R.; Mitri, S.; Ciccarese, D.; Or, D.; van der Meer, J.R.; Johnson, D.R. Bridging the Holistic-Reductionist Divide in Microbial Ecology. mSystems 2019, 4, e00265-18.
- Gilmore, S.P.; Lankiewicz, T.S.; Wilken, S.E.; Brown, J.L.; Sexton, J.A.; Henske, J.K.; Theodorou, M.K.; Valentine, D.L.; O’Malley, M.A. Top-Down Enrichment Guides in Formation of Synthetic Microbial Consortia for Biomass Degradation. ACS Synth. Biol. 2019, 8, 2174–2185.
- Qin, Y.; Druzhinina, I.S.; Pan, X.; Yuan, Z. Microbially Mediated Plant Salt Tolerance and Microbiome-Based Solutions for Saline Agriculture. Biotechnol. Adv. 2016, 34, 1245–1259.
- Niu, B.; Paulson, J.N.; Zheng, X.; Kolter, R. Simplified and Representative Bacterial Community of Maize Roots. Proc. Natl. Acad. Sci. USA 2017, 114, E2450–E2459.
- Kaminsky, L.M.; Trexler, R.V.; Malik, R.J.; Hockett, K.L.; Bell, T.H. The Inherent Conflicts in Developing Soil Microbial Inoculants. Trends Biotechnol. 2019, 37, 140–151.
- Burke, C.; Steinberg, P.; Rusch, D.; Kjelleberg, S.; Thomas, T. Bacterial Community Assembly Based on Functional Genes Rather than Species. Proc. Natl. Acad. Sci. USA 2011, 108, 14288–14293.
- Tsolakidou, M.-D.; Stringlis, I.A.; Fanega-Sleziak, N.; Papageorgiou, S.; Tsalakou, A.; Pantelides, I.S. Rhizosphere-Enriched Microbes as a Pool to Design Synthetic Communities for Reproducible Beneficial Outputs. FEMS Microbiol. Ecol. 2019, 95, fiz138.
- Minchev, Z.; Kostenko, O.; Soler, R.; Pozo, M.J. Microbial Consortia for Effective Biocontrol of Root and Foliar Diseases in Tomato. Front. Plant Sci. 2021, 12, 756368.
- Timofeeva, A.M.; Galyamova, M.R.; Sedykh, S.E. Bacterial Siderophores: Classification, Biosynthesis, Perspectives of Use in Agriculture. Plants 2022, 11, 3065.
- Timofeeva, A.; Galyamova, M.; Sedykh, S. Prospects for Using Phosphate-Solubilizing Microorganisms as Natural Fertilizers in Agriculture. Plants 2022, 11, 2119.
- Nadeem, S.M.; Ahmad, M.; Zahir, Z.A.; Javaid, A.; Ashraf, M. The Role of Mycorrhizae and Plant Growth Promoting Rhizobacteria (PGPR) in Improving Crop Productivity under Stressful Environments. Biotechnol. Adv. 2014, 32, 429–448.
- Zimmer, S.; Messmer, M.; Haase, T.; Piepho, H.-P.; Mindermann, A.; Schulz, H.; Habekuß, A.; Ordon, F.; Wilbois, K.-P.; Heß, J. Effects of Soybean Variety and Bradyrhizobium Strains on Yield, Protein Content and Biological Nitrogen Fixation under Cool Growing Conditions in Germany. Eur. J. Agron. 2016, 72, 38–46.
- Armanhi, J.S.L.; De Souza, R.S.C.; Damasceno, N.D.B.; De Araujo, L.M.; Imperial, J.; Arruda, P. A Community-Based Culture Collection for Targeting Novel Plant Growth-Promoting Bacteria from the Sugarcane Microbiome. Front. Plant Sci. 2018, 8, 2191.
- Toju, H.; Peay, K.G.; Yamamichi, M.; Narisawa, K.; Hiruma, K.; Naito, K.; Fukuda, S.; Ushio, M.; Nakaoka, S.; Onoda, Y.; et al. Core Microbiomes for Sustainable Agroecosystems. Nat. Plants 2018, 4, 247–257.
- Fahad, S.; Bajwa, A.A.; Nazir, U.; Anjum, S.A.; Farooq, A.; Zohaib, A.; Sadia, S.; Nasim, W.; Adkins, S.; Saud, S.; et al. Crop Production under Drought and Heat Stress: Plant Responses and Management Options. Front. Plant Sci. 2017, 8, 1147.
- Niu, X.; Song, L.; Xiao, Y.; Ge, W. Drought-Tolerant Plant Growth-Promoting Rhizobacteria Associated with Foxtail Millet in a Semi-Arid Agroecosystem and Their Potential in Alleviating Drought Stress. Front. Microbiol. 2018, 8, 2580.
- Paredes-Páliz, K.; Rodríguez-Vázquez, R.; Duarte, B.; Caviedes, M.A.; Mateos-Naranjo, E.; Redondo-Gómez, S.; Caçador, M.I.; Rodríguez-Llorente, I.D.; Pajuelo, E. Investigating the Mechanisms Underlying Phytoprotection by Plant Growth-promoting Rhizobacteria in Spartina Densiflora under Metal Stress. Plant Biol. 2018, 20, 497–506.
- Kasim, W.A.; Gaafar, R.M.; Abou-Ali, R.M.; Omar, M.N.; Hewait, H.M. Effect of Biofilm Forming Plant Growth Promoting Rhizobacteria on Salinity Tolerance in Barley. Ann. Agric. Sci. 2016, 61, 217–227.
- Xu, N.; Yang, L.; Fan, Y.; Yang, J.; Yue, D.; Liang, Y.; Price, B.; Cohen, S.; Huang, T. YouTube-VOS: Sequence-to-Sequence Video Object Segmentation; Springer: Berlin, Germany, 2018; pp. 603–619.
- Shaheen, R.; Svensson, B.; Andersson, M.A.; Christiansson, A.; Salkinoja-Salonen, M. Persistence Strategies of Bacillus Cereus Spores Isolated from Dairy Silo Tanks. Food Microbiol. 2010, 27, 347–355.
- Poli, A.; Anzelmo, G.; Nicolaus, B. Bacterial Exopolysaccharides from Extreme Marine Habitats: Production, Characterization and Biological Activities. Mar. Drugs 2010, 8, 1779–1802.
- Nunkaew, T.; Kantachote, D.; Nitoda, T.; Kanzaki, H.; Ritchie, R.J. Characterization of Exopolymeric Substances from Selected Rhodopseudomonas Palustris Strains and Their Ability to Adsorb Sodium Ions. Carbohydr. Polym. 2015, 115, 334–341.
- Ashraf, M.; Hasnain, S.; Berge, O.; Mahmood, T. Inoculating Wheat Seedlings with Exopolysaccharide-Producing Bacteria Restricts Sodium Uptake and Stimulates Plant Growth under Salt Stress. Biol. Fertil. Soils 2004, 40, 157–162.
- Zörb, C.; Geilfus, C.-M.; Dietz, K.-J. Salinity and Crop Yield. Plant Biol. 2019, 21, 31–38.
- Kumar, A.; Singh, S.; Mukherjee, A.; Rastogi, R.P.; Verma, J.P. Salt-Tolerant Plant Growth-Promoting Bacillus Pumilus Strain JPVS11 to Enhance Plant Growth Attributes of Rice and Improve Soil Health under Salinity Stress. Microbiol. Res. 2021, 242, 126616.
- Kumar Arora, N.; Fatima, T.; Mishra, J.; Mishra, I.; Verma, S.; Verma, R.; Verma, M.; Bhattacharya, A.; Verma, P.; Mishra, P.; et al. Halo-Tolerant Plant Growth Promoting Rhizobacteria for Improving Productivity and Remediation of Saline Soils. J. Adv. Res. 2020, 26, 69–82.
- Turan, M.; Gulluce, M.; Şahin, F. Effects of Plant-Growth-Promoting Rhizobacteria on Yield, Growth, and Some Physiological Characteristics of Wheat and Barley Plants. Commun. Soil Sci. Plant Anal. 2012, 43, 1658–1673.
- Mishra, P.K.; Bisht, S.C.; Ruwari, P.; Selvakumar, G.; Joshi, G.K.; Bisht, J.K.; Bhatt, J.C.; Gupta, H.S. Alleviation of Cold Stress in Inoculated Wheat (Triticum aestivum L.) Seedlings with Psychrotolerant Pseudomonads from NW Himalayas. Arch. Microbiol. 2011, 193, 497–513.
- Wu, H.; Gu, Q.; Xie, Y.; Lou, Z.; Xue, P.; Fang, L.; Yu, C.; Jia, D.; Huang, G.; Zhu, B.; et al. Cold-adapted Bacilli Isolated from the Qinghai–Tibetan Plateau Are Able to Promote Plant Growth in Extreme Environments. Environ. Microbiol. 2019, 21, 3505–3526.
- Knief, C.; Delmotte, N.; Chaffron, S.; Stark, M.; Innerebner, G.; Wassmann, R.; von Mering, C.; Vorholt, J.A. Metaproteogenomic Analysis of Microbial Communities in the Phyllosphere and Rhizosphere of Rice. ISME J. 2012, 6, 1378–1390.
- Sessitsch, A.; Hardoim, P.; Döring, J.; Weilharter, A.; Krause, A.; Woyke, T.; Mitter, B.; Hauberg-Lotte, L.; Friedrich, F.; Rahalkar, M.; et al. Functional Characteristics of an Endophyte Community Colonizing Rice Roots as Revealed by Metagenomic Analysis. Mol. Plant-Microbe Interact. 2012, 25, 28–36.
- Bakker, P.A.H.M.; Berendsen, R.L.; Doornbos, R.F.; Wintermans, P.C.A.; Pieterse, C.M.J. The Rhizosphere Revisited: Root Microbiomics. Front. Plant Sci. 2013, 4, 165.
- Forchetti, G.; Masciarelli, O.; Alemano, S.; Alvarez, D.; Abdala, G. Endophytic Bacteria in Sunflower (Helianthus annuus L.): Isolation, Characterization, and Production of Jasmonates and Abscisic Acid in Culture Medium. Appl. Microbiol. Biotechnol. 2007, 76, 1145–1152.
- Lakshmanan, V.; Selvaraj, G.; Bais, H.P. Functional Soil Microbiome: Belowground Solutions to an Aboveground Problem. Plant Physiol. 2014, 166, 689–700.
- Schlaeppi, K.; Bulgarelli, D. The Plant Microbiome at Work. Mol. Plant-Microbe Interact. 2015, 28, 212–217.
- Jansson, J.K.; Hofmockel, K.S. The Soil Microbiome—From Metagenomics to Metaphenomics. Curr. Opin. Microbiol. 2018, 43, 162–168.
- Zhou, J.; He, Z.; Yang, Y.; Deng, Y.; Tringe, S.G.; Alvarez-Cohen, L. High-Throughput Metagenomic Technologies for Complex Microbial Community Analysis: Open and Closed Formats. MBio 2015, 6, e02288-44.
- Bulgarelli, D.; Garrido-Oter, R.; Münch, P.C.; Weiman, A.; Dröge, J.; Pan, Y.; McHardy, A.C.; Schulze-Lefert, P. Structure and Function of the Bacterial Root Microbiota in Wild and Domesticated Barley. Cell Host Microbe 2015, 17, 392–403.
- Peng, J.; Wegner, C.-E.; Bei, Q.; Liu, P.; Liesack, W. Metatranscriptomics Reveals a Differential Temperature Effect on the Structural and Functional Organization of the Anaerobic Food Web in Rice Field Soil. Microbiome 2018, 6, 169.
- Roy Chowdhury, T.; Lee, J.-Y.; Bottos, E.M.; Brislawn, C.J.; White, R.A.; Bramer, L.M.; Brown, J.; Zucker, J.D.; Kim, Y.-M.; Jumpponen, A.; et al. Metaphenomic Responses of a Native Prairie Soil Microbiome to Moisture Perturbations. mSystems 2019, 4, e00061-19.
- Guttman, D.S.; McHardy, A.C.; Schulze-Lefert, P. Microbial Genome-Enabled Insights into Plant–Microorganism Interactions. Nat. Rev. Genet. 2014, 15, 797–813.
- Kleiner, M.; Thorson, E.; Sharp, C.E.; Dong, X.; Liu, D.; Li, C.; Strous, M. Assessing Species Biomass Contributions in Microbial Communities via Metaproteomics. Nat. Commun. 2017, 8, 1558.
- Neurohr, G.E.; Amon, A. Relevance and Regulation of Cell Density. Trends Cell Biol. 2020, 30, 213–225.
- Milo, R. What Is the Total Number of Protein Molecules per Cell Volume? A Call to Rethink Some Published Values. BioEssays 2013, 35, 1050–1055.
- Kleiner, M. Metaproteomics: Much More than Measuring Gene Expression in Microbial Communities. mSystems 2019, 4, e00115-19.
- Thiele, I.; Palsson, B.Ø. A Protocol for Generating a High-Quality Genome-Scale Metabolic Reconstruction. Nat. Protoc. 2010, 5, 93–121.
- Shoaie, S.; Ghaffari, P.; Kovatcheva-Datchary, P.; Mardinoglu, A.; Sen, P.; Pujos-Guillot, E.; de Wouters, T.; Juste, C.; Rizkalla, S.; Chilloux, J.; et al. Quantifying Diet-Induced Metabolic Changes of the Human Gut Microbiome. Cell Metab. 2015, 22, 320–331.
- Lander, E.S.; Waterman, M.S. Genomic Mapping by Fingerprinting Random Clones: A Mathematical Analysis. Genomics 1988, 2, 231–239.
More