Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Wojciech Grzegorz Wiese and Version 2 by Sirius Huang.
Blood malignancies remain a therapeutic challenge despite the development of numerous treatment strategies. The phosphatidylinositol-3 kinase (PI3K)/protein kinase B/mammalian target of rapamycin (PI3K/Akt/mTOR) signaling pathway plays a central role in regulating many cellular functions, including cell cycle, proliferation, quiescence, and longevity. Therefore, dysregulation of this pathway is a characteristic feature of carcinogenesis.
- PI3K/Akt/mTOR pathway
- leukemia
- PI3K inhibitors
- mTOR inhibitors
- dual PI3K/mTOR inhibitors
- Akt inhibitors
1. Introduction
Blood malignancies are a heterogeneous group of neoplasms arising due to the disruption of normal hematopoiesis [1][2][1,2]. They are among the most common cancer types, accounting for 6.5% of all cancers around the world [3]. Hematologic malignancies are generally classified into leukemias, multiple myeloma (MM), non-Hodgkin lymphomas (NHLs), and Hodgkin lymphoma (HL) [4]. Each disease is characterized by different morphological features, prognosis, and treatment regimens [5].
The PI3K/Akt/mTOR pathway is one of the most important signaling pathways that regulates cell growth and proliferation. PI3K/Akt/mTOR activation is important for leukemogenesis and it is associated with an unfavorable prognosis. The pathway is apparently dysregulated in numerous human malignancies such as respiratory tumors, digestive system tumors, kidney cancer, skin cancer, breast, and ovarian cancer, to name a few [6][7][6,7]. The activation of the PI3K/Akt/mTOR axis has been detected in hematologic malignancies, including ALL, CLL, AML, and CML. PI3K/Akt/mTOR activation is important for leukemogenesis and it is associated with unfavorable prognosis. Inhibition of this pathway through specific inhibitors results in reduced leukemic cell proliferation [8][9][8,9]. In T-ALL, mutations and deletions leading to the phosphatase and tensin homolog (PTEN) loss are the most common reasons for the upregulation of the PI3K/Akt/mTOR signaling, as PTEN is a major negative regulator of this pathway. PTEN loss contributes to increased cell proliferation and chemoresistance in AML, ALL, and CML [10]. The PI3K/Akt/mTOR pathway is also known to play an important role in CLL, where it promotes autophagy and thus contributes to improved cell survival [11]. Moreover, the PI3K/Akt/mTOR axis is associated with several mutations in hematological malignancies. For instance, FLT3 mutations in AML promote proliferation through mTOR signaling, whereas BCR-ABL kinase in CML activates the PI3K/Akt/mTOR pathway by binding to the p85 PI3K regulatory subunit [11][12][11,12].
2. The PI3K/Akt/mTOR Signaling Pathway
PI3K is a lipid kinase family that phosphorylates inositol’s 3′-OH group in phospholipids on the plasma membrane [13]. Human cells contain three classes of PI3Ks. Class I PI3Ks consist of a catalytic isoform and a regulatory subunit which mediate the kinase activity. This class is divided into two subclasses—IA, activated by receptor tyrosine kinases (RTKs), and IB, activated by G protein-coupled receptors (GPCR) [14][15][14,15]. Genes PIK3CA, PIK3CB, and PI3KCD encode the class IA catalytic subunit isoforms p110α, p110β, and p110δ, respectively. These catalytic isoforms can form heterodimers with any of the regulating subunit isoforms, p85α, p55α or p50α (PIK3R1), p85β (PIK3R2), and p55γ (PIK3R3), all of which are p85α splicing variants. PIK3CG encodes the class IB p110γ catalytic subunit, which forms heterodimers with regulatory isoforms—p101 or p87, encoded, respectively, by PIK3R5 and PIK3R6. Interestingly, while p110α and p110β are ubiquitously expressed in all tissues, p110δ and p110γ expression seems to be highly restricted to leukocytes and the hematopoietic system [14][16][17][18][14,16,17,18]. Class II PI3Ks contain only the catalytic subunit. This subunit has three isoforms—PI3K-C2α, PI3K-C2β, and PI3K-C2γ, encoded, respectively, by PIK3C2A, PIK3C2B, and PIK3C2G. Class III PI3Ks consist of Vps34 (vacuolar protein sorting 34), which is encoded by PIK3C3 [15][19][15,19] (Table 1).Table 1. The table presents the fundamental classes of the phosphatidylinositol-3 kinase (PI3K), along with their subclasses and isoforms. The table also includes genes encoding a given PI3K class.
| PI3K Class | Subunits | Isoforms | Encoding Gene |
|---|---|---|---|
| IA | Catalytic subunits + regulating subunit isoform |
p110α, p110β, p110δ | PIK3CA, PIK3CB, PI3KCD |
| IB | - | p110γ | PIK3CG |
| II | Only the catalytic subunit | PI3K-C2α, PI3K-C2β, PI3K-C2γ | PIK3C2A, PIK3C2B, PIK3C2G |
| III | Vps34 | - | PIK3C3 |
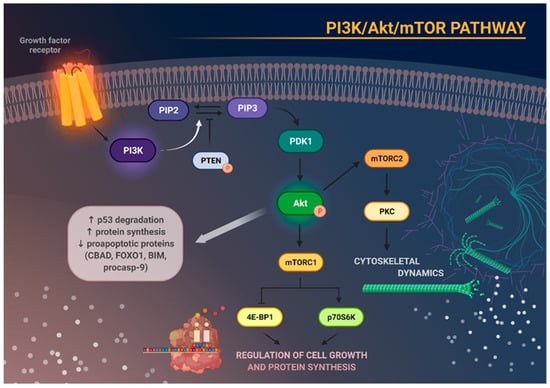
Figure 1. Schematic representation of the main molecular consequences of phosphoinositide 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway activation. Stimulation of growth factor receptor activates PI3K, which in turn causes phosphorylation of phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-triphosphate (PIP3). This process can be reversed by phosphatase and tensin homolog (PTEN). Increased activity of PIP3 results in recruitment of phosphoinositide-dependent kinase 1 (PDK1), which subsequently phosphorylates and activates Akt. Activation of Akt degrades p53, increases protein synthesis, and inhibits the activity of several proapoptotic proteins. Akt, through activation of mTOR, enables the formation of the two complexes—mTORC1 and mTORC2. mTORC1 phosphorylates and activates eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) and p70S6 kinase (p70S6K), which are responsible for cell growth and protein synthesis. mTORC2, on the other hand, regulates cytoskeletal architecture through protein kinase C (PKC) phosphorylation.
3. Application of PI3K/Akt/mTOR Pathway Inhibitors in Blood Malignancies
Depending on the mechanism of action, PI3K/Akt/mTOR pathway inhibitors are divided into PI3K kinase inhibitors, Akt kinase inhibitors, and mTOR kinase inhibitors. There are also dual inhibitors that inhibit both PI3K and mTOR kinase. Nowadays, the Food and Drug Administration (FDA) has approved two inhibitors of the PI3K/Akt/mTOR pathway for leukemia treatment; these are Idelalisib and Duvelisib, for the treatment of CLL.
3.1. PI3K Inhibitors
PI3K inhibitors are classified as isoform-specific, pan-PI3K (targeting all four isoforms, α, β, δ, and γ of class I PI3K), and dual PI3K/mTOR inhibitors. Among all pan-PI3K inhibitors, only Copanlisib has been approved by the FDA. The other known pan-PI3K inhibitors are Buparlisib and ZSTK474. Idelalisib and Umbralisib inhibit the δ isoform, whereas Duvelisib inhibits the δ/γ isoforms of PI3K [27][64].
Idelalisib is a PI3Kδ inhibitor approved in 2014 by the FDA. It is used for treating CLL in combination with Rituximab in disease resistance or relapse after at least one line of therapy. Idelalisib is the first-line agent for patients with del17p or TP53 mutations ineligible for immunochemotherapy. Idelalisib is also approved in follicular lymphoma (FL) after two lines of therapy [28][65]. Despite the FDA approval of Idelalisib for treating CLL and FL, studies on the efficacy of this drug combined with other drugs are constantly underway. The study with Idelalisib in combination with Rituximab presents increased PFS (progression-free survival) in patients with relapsed CLL, compared to placebo plus Rituximab administration. Median PFS in the Idelalisib plus Rituximab (I–R) group was 20.3 months after a median follow-up time of 18 months. Patients who received I–R in the main study and continued treatment with Idelalisib alone had a median PFS of 20.3 months (95% CI, 17.3–26.3 months), and an overall response rate (ORR) of 85.5%. Median OS (overall survival) was 40.6 months and 34.6 months for patients assigned randomly to I–R and placebo plus Rituximab groups, respectively. Treatment with Idelalisib did not increase the incidence of elevated liver aminotransferases; however, it increased the incidence of diarrhea, colitis, and pneumonia [29][66]. On the other hand, another study revealed that treatment with I–R proved less effective for patients with relapsed/refractory CLL than monotherapy with Acalabrutinib—a Bruton’s tyrosine kinase inhibitor. Median PFS and estimated 12-month PFS were significantly longer in the case of Acalabrutinib monotherapy compared with I–R or Bendamustine plus Rituximab (B–R) therapy. Serious adverse events (Aes) occurred in 29% of patients treated with Acalabrutinib monotherapy, 56% treated with I–R, and 26% treated with B–R. Deaths occurred in 10%, 11%, and 14% of patients receiving Acalabrutinib monotherapy, I–R, and B–R, respectively [30][67]. Altogether, it has been concluded that I–R therapy is less effective and has more side effects in relapsed/refractory CLL than Acalabrutinib.
Duvelisib is an orally available dual inhibitor of PI3K-δ approved by the FDA for use in relapsed/refractory CLL or small lymphocytic lymphoma (SLL) after at least two prior therapies [31][68]. In a Phase I study, Duvelisib monotherapy in patients with relapsed/refractory CLL resulted in an ORR of 56% [32][69]. Studies on the efficacy of Duvelisib in combination with other drugs are ongoing. One study analyzed the efficacy and toxicity of Duvelisib in combination with Rituximab (ARM 1) and Duvelisib with Bendamustine and Rituximab (ARM 2) in patients with relapsed/refractory non-Hodgkin lymphoma (NHL) and CLL. Patients with CLL had better treatment outcomes in ARM 1 (ARM 1—partial response (PR) 88.9%, ORR 88.9% vs. ARM 2—PR 50%, ORR 75.0%). Duvelisib in combination with Rituximab or Bendamustine and Rituximab did not increase treatment toxicity [33][70]. The combination of Duvelisib with Rituximab may become a new, promising treatment strategy. However, more studies are needed to confirm their combined efficacy.
The other Inhibitor of pan-PI3K is Buparlisib. Buparlisib therapy in patients with refractory/relapsed CLL resulted in an ORR of 46% with a median duration of response of 15.5 months. The most common side effects after Buparlisib administration were hyperglycemia, fatigue, anxiety, and gastrointestinal toxicities [34][35][71,72]. However, in a Phase I trial with patients with refractory/relapsed AML and refractory/relapsed ALL, Buparlisib was less effective, with a median survival time of 75 days [35][72]. On the other hand, Buparlisib was more cytotoxic against B-CLL cells than Idelalisib in in vitro studies [36][73].
The next studied PI3K inhibitor is Umbralisib. Umbralisib inhibits PI3Kδ isoform as well as casein kinase-1ε. Patients with CLL have reached a PFS of 23.5 months with Umbralisib therapy [37][74]. Therapy based on Umbralisib plus Ibrutinib in patients with relapsed/refractory CLL achieved an ORR of 90%, and PR/PR with lymphocytosis accounted for 29% [38][75]. Every AE was graded according to the Common Terminology Criteria for Adverse Events grade. The most common Aes were diarrhea, nausea, and fatigue, and the most common grade 3 or higher AEs were anemia and thrombocytopenia. The most common AEs leading to early discontinuation of therapy were rash, arthralgia, and atrial fibrillation [37][39][40][74,76,77].
Another pan-PI3K inhibitor applied in preclinical studies is ZSTK474. ZSTK474 inhibited the cell growth of AML and ALL in in vitro studies [41][42][78,79]. The inhibition of cell growth was concentration-dependent. ZSTK474 monotherapy induced apoptosis both in AML and ALL cells. Upon combination of ZSTK474 with the extracellular signal-regulated kinase 1/2 (ERK1/2) inhibitor AZD0364, the apoptosis of ALL and AML cells was increased; this was associated with the induction of oxidative stress and cellular antioxidant defense mechanisms [42][79]. Moreover, ZSTK474 exhibited cytotoxic effects against T-ALL and B-ALL cells [43][44][80,81]. In a separate study, ZSTK474 reduced CML cells viability and proliferation by inducing cell cycle arrest at the G1 phase. Further, the combination of ZSTK474 with Imatinib showed a synergistic effect, improving the effectiveness of therapy also in the multidrug-resistant counterpart cells [45][82]. Another in vitro study tested the efficacy of ZSTK474 in combination with Imatinib, Nilotinib, and the BCR-ABL inhibitor GZD824 on Philadelphia chromosome-positive B-ALL cells. The combination of these drugs decreased cell viability and induced apoptosis and autophagy [46][83].
3.2. mTOR Inhibitors
The next group of drugs described herein is mTOR inhibitors, already used in immunosuppression and cancer treatment. The first generation of mTOR inhibitors comprises natural rapamycin (Sirolimus) and its synthetic analogs, known as rapalogists. These inhibitors bind to the FKBP-rapamycin-binding (FRB) domain. Second-generation ATP-competitive mTOR inhibitors (TOR-Ki) can effectively block both mTORC1 and mTORC2 by binding to the ATP-binding pocket of the kinase catalytic domain (KIN). The third generation of mTOR inhibitors is called RapaLinks or bi-steric mTORC1 inhibitors; these are made by connecting rapamycin and TOR-Ki. This generation embraces the action of both first- and second-generation mTOR inhibitors [47][48][84,85].
Everolimus is an oral mTOR kinase inhibitor used to prevent the rejection of transplanted organs and treat breast cancer, pancreatic-derived neuroendocrine tumors, and renal cell carcinoma, among others [49][86]. In a randomized trial in patients with AML, Everolimus did not increase relapse-free survival, the cumulative incidence of relapse, or OS. The study randomized patients to receive Everolimus between consolidation chemotherapy courses. The study terminated due to excess mortality in the Everolimus arm, without any evidence of beneficial disease control [50][87]. In a study of childhood patients with relapsed ALL, therapy with Everolimus combined with Vincristine, Prednisone, Pegaspargase, and Doxorubicin led to complete remission in 19 of 22 consecutive patients. Complete remission occurred in all six patients with a known KMT2A or iAMP21 rearrangement. The combination of Everolimus with the drugs mentioned above was well tolerated [51][88]. Everolimus combined with HyperCVAD (Cyclophosphamide + Vincristine + Doxorubicin + Dexamethasone) chemotherapy resulted in better treatment outcomes than first-line HyperCVAD alone—partial response (PR) or complete response (CR) of 63.6% and 53.3%, respectively. The results were not statistically significant, however, and additional studies on a larger group of patients are needed to confirm the effectiveness of the therapy. The therapy of Everolimus plus HyperCVAD was also well tolerated [52][89].
Rapamycin (Sirolimus—trade name) is an mTOR inhibitor that was approved by the FDA in 1999 for the prevention of kidney transplant rejection. Rapamycin is a macrocyclic lactone produced by Streptomyces hygroscopicus, which was isolated from soil samples in the late 1960s. Rapamycin or its rapalogues are also applied in the prevention of restenosis after coronary angioplasty and used in oncology—the FDA approved the use of rapamycin to treat patients with pancreatic cancer in 2011 [53][54][90,91]. In a clinical trial in high-risk AML patients treated with Sirolimus in combination with MEC (Mitoxantrone + Etoposide + Cytarabine), the ORR was 47% (CR 33%, complete remission with incomplete hematologic recovery 2%, PR 12%). Moreover, ORR was not significantly different between participants with and without baseline mTORC1 activity (52% vs. 40%, respectively). Sirolimus therapy together with MEC was well tolerated [55][92]. In another Phase II study in patients with relapsed/refractory AML, Sirolimus in combination with MEC had an ORR of 16%. The study also examined the efficacy of the Carboplatin + Topotecan and Alvocidib + Cytarabine + Mitoxantrone scheme. The ORR accounted for 14% and 28%, respectively [56][93].
Temsirolimus is an mTOR inhibitor that was approved by the FDA in 2007 for the treatment of advanced renal cell carcinoma [47][84]. The combination of Temsirolimus with a UKALL R3 reinduction chemotherapy regimen (Dexamethasone + Vincristine + Mitoxantrone + Pegaspargase + intrathecal Methotrexate) was investigated in childhood relapsed/refractory ALL patients. In the study, 46.6% of patients achieved remission, while 20% had residual disease of <0.01%. In therapy-related AEs, 73% of the children studied developed neutropenic fever and 53% of patients had a documented grade 3 or 4 infection and one grade 5 bacterial sepsis [57][94]. Another study examined the therapy of Temsirolimus combined with Clofarabine in patients with AML. The treatment resulted in an ORR of 21%, of which 8% achieved CR. Median disease-free survival was 3.5 months, and median OS was 4 months [58][95].
RMC-4627 is a novel bi-steric mTORC1-selective inhibitor; at this moment it is undergoing preclinical studies. In in vitro research, RMC-4627 demonstrated strong and selective inhibition of 4E-BP1 phosphorylation specifically within B-ALL cell lines, while mTORC2 activity was unaffected. RMC-4627 reduced proliferation, decreased survival, and significantly increased the efficacy and tolerability of Dasatinib in a Ph+ B-ALL xenograft model [48][59][85,96]. It is worth adding that the first clinical candidate in the class of bi-steric mTORC1 inhibitor (RMC-5552) is undergoing clinical trials in solid tumors (NCT04774952) [60][97].
3.3. Dual PI3K/mTOR Inhibitors
Dual PI3K/mTOR inhibitors are another group of potential drugs undergoing extensive investigation. Dual PI3K/mTOR inhibitors can completely suppress the aberrant activation of the PI3K/Akt/mTOR signaling pathway and prevent the compensatory activation of the Akt/mTOR pathway; this can lead to improved treatment outcomes [61][98].
Gedatolisib was found to reduce the number of ALL cells in the spleen by an average of 91.8% in patient-derived xenograft mouse models of childhood Ph-like ALL. In a xenograft mice with cytokine receptor 2 (CRLF2)/JAK Ph-like ALL mutations, Gedatolisib therapy reduced the viability of ALL cells by 92.2%. Gedatolisib also inhibited ALL proliferation in ABL/platelet-derived growth factor receptor (PDGFR) mutant models with a mean reduction of 66.9%. The high efficacy of Gedatolisib correlated with inhibition of phosphorylated ribosomal protein S6 (pS6) and 4E-BP1 in Ph-like ALL models [62][99]. Gedatolisib significantly prolonged survival of mice in a xenograft model of Sorafenib-resistant AML [63][100]. Furthermore, Gedatolisib treatment led to marked inhibition of T-ALL growth compared to vehicle treatment, as well as delayed tumor growth in all treated mice [64][101].
Imidazoquinoline derivative BEZ235 is a dual PI3K/mTOR inhibitor. BEZ235 is currently undergoing Phase I clinical trials. In one of the studies, in patients with refractory or relapsed leukemia, the response was observed in 2 of 10 patients with BCP-ALL and 1 of 1 patient with T-ALL. In contrast, there was no response in any patient with AML (n = 12) or CML (n = 1). The response to BEZ235 treatment was uncorrelated with the level of PI3K signaling markers. BEZ235 therapy was mainly associated with gastrointestinal-related toxicity [65][102]. In vitro, BEZ235 reduced viability, induced G0/G1 arrest, and increased apoptosis of AML cells [66][67][103,104]. In xenograft models of AML MLL-AF9+/FLT3-ITD+, BEZ235 therapy resulted in delayed tumor progression and prolonged survival [68][105]. It was also found that BEZ235 inhibited AML cell migration and sensitized cells to Vincristine and Adriamycin [67][104]. Furthermore, in either in vitro or in vivo models of T-ALL, inhibition of PI3K/mTOR with BEZ235 enhanced the antileukemic effect of Dexamethasone [69][106]. BEZ235 also inhibited proliferation, induced apoptosis, and activated autophagy in CML cells in both cellular and xenograft models [70][71][107,108]. Moreover, BEZ235 increased the sensitivity of CML cells to Imatinib [71][108]. Another study showed that BEZ235 inhibited proliferation of adult T-cell leukemia (ATL) cells in in vivo models. In CML cells treated with BEZ235, PI3K/Akt/mTOR activity and the levels of the antiapoptotic protein Bcl-2 decreased, while the levels of the proapoptotic protein Bax increased [66][72][103,109]. BEZ235 also offers the opportunity to overcome resistance to Venetoclax, a selective Bcl-2 inhibitor. Venetoclax has significantly enhanced the treatment options available to patients with refractory and relapsed blood cancers, including those with AML. Venetoclax has fewer side effects, which makes it more effective for elderly patients. Numerous studies have demonstrated that the two major antiapoptotic Bcl-2 family proteins, namely, Bcl-XL and MCL-1, serve as the primary factors that determine resistance to Venetoclax. Venetoclax has a high binding specificity for Bcl-2; thus, the relative expression levels of Bcl-2 proteins may be a determinant of Venetoclax resistance. Combining Venetoclax with other targeted drugs such as BEZ235, a dual PI3K/mTOR inhibitor, offers a chance to circumvent resistance to Venetoclax [9][11][73][74][9,11,110,111].
3.4. Akt Inhibitors
GSK2141795 is an ATP-competitive, fully reversible pan-Akt kinase inhibitor. GSK2141795 inhibited neoplastic cell proliferation with activated Akt pathway in vitro and in vivo [75][112]. A Phase II study enrolled patients with relapsed/refractory AML with an RAS mutation, in which GSK2141795 therapy was combined with Trametinib (GSK1120212)—a dual-specificity mitogen-activated protein kinase kinase 1 and 2 inhibitor (MEK1 and MEK2). No patient achieved CR and CR with incomplete recovery of platelets due to therapy, and the study was closed early due to lack of clinical activity. The median OS amounted to 3 months. The most common AEs were diarrhea, maculopapular rash, and mucositis. Serious AEs (grade 3–4) were observed in 39% of patients, the most frequent being rash, mucositis, and diarrhea [76][113].