1. Focus on the Most Used Animal Models in Corneal Pathologies
Corneal diseases are the fifth leading cause of blindness worldwide, after cataracts, refractive errors, glaucoma, and age-related macular degeneration
[12][1]. Thus, this section brings together the different corneal pathologies with the most relevant and widely used animal models. The main objective is to highlight the evidence for the choice of animals to better understand and cure these corneal diseases.
2. Dry Eye Diseases
2.1. Pathology
Dry eye disease (DED) is the most frequent disorder in ophthalmology. DED is a multifactorial tear film and ocular surface disease causing both objective and subjective symptoms
[140[2][3],
141], mainly due to insufficient tear production, excessive tear evaporation, or goblet cell loss
[142,143][4][5]. Destabilization of the ocular surface mucosa, composed of mucins, is one of the main causes of dry eye
[55,59,144][6][7][8]. Mucins are secreted by conjunctival goblet cells and lacrimal glands and are expressed at the membrane level of corneal and conjunctival epithelial cells. They are essential for maintaining the wettability of the ocular surface and thus contribute to the dynamics, stability, and osmolarity of the tear film. Thus, alterations in mucin expression can lead to increased water loss from tears, ultimately contributing to tear hyperosmolarity, which has been associated with ocular surface inflammation
[59,144][7][8]. The glands involved in the tear film secretion are the lacrimal glands, which produce and remove the aqueous layer, while the Meibomian glands produce the lipid layer (
Figure 2). This lipid layer is then spread over the tear film with each blink to stabilize it
[59][7]. Therefore, tear film secretions from the lacrimal and the Meibomian gland contribute to the mucin content of the ocular surface. On the one hand, objective signs are tear film instability with the potential for ocular surface damage, increased osmolarity of the tear film, mucus discharge, increased frequency of blinking-tearing, and ocular surface inflammation
[140,141,143][2][3][5]. On the other hand, subjective symptoms include visual disturbances (i.e., blurred) and discomfort (sensations of dryness or foreign body, pain, irritation, redness, burning, itching, sensitivity to light, and intolerance to contact lenses)
[140,141,145][2][3][9]. Unlike objective signs, which are quantifiable and can also be assessed in animal models, subjective symptoms are evaluated using questionnaires and visual function tests (visual acuity at high and low contrast, dynamic visual acuity, and contrast sensitivity) in the patient
[62,76,140,141,143,145,146][2][3][5][9][10][11][12]. Numerous studies have shown that psychological effects (depression, anxiety, feelings of happiness, etc.), personality traits, and the patient’s sensitivity to pain influence subjective symptoms of DED
[140][2], making it difficult to use and interpret these symptoms in animal models. As an example, there is evidence of a relationship between patients’ subjective happiness and reported dry eye symptoms
[147][13]. One study showed that a more enriched environment (e.g., exercise, well-being, sensory, cognitive, and social stimuli) in a stress-induced DED mouse model compared to a standard environment provides an effective intervention to prevent and attenuate decreased tear secretion in DED
[148][14]. Behavioral observations can also be used to assess visual acuity in DED animal models using rodents or nonhuman primates
[91,149][15][16]. Moreover, several factors can affect the DED severity, including autoimmune diseases and hormonal changes that play an important role in regulating tear production by the lacrimal gland
[150][17], environmental surroundings (pollution), daily activities (watching TV, reading, mobile devices or computer), contact lens use, anatomical features, chronic inflammation, infections, and iatrogenic factors, such as medications or surgery
[143,151,152][5][18][19]. The global prevalence of DED is from 5% to 50% with a higher rate in women than in men
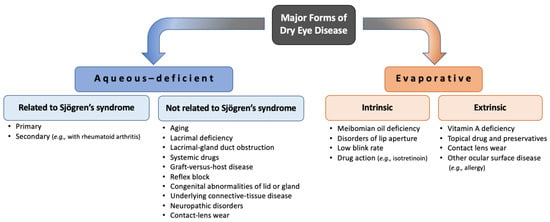
[153][20]. In 2017, the report from the Tear Film and Ocular Surface Society International Dry Eye Workshop II defined and updated the classification of DED into two main classes: aqueous-deficient dry eye and evaporative dry eye
[154][21]. Although aqueous-deficient dry eye and evaporative dry eye show similar signs of reduced stability and increased osmolarity of the tear film, aqueous-deficient dry eye refers primarily to a failure of lacrimal secretion and evaporative dry eye is due to excessive water loss from the exposed ocular surface in the presence of normal tear secretory function
[155][22]. It is important for clinicians and researchers to consider both forms of dry eye when diagnosing, treating, monitoring, and establishing animal models of DED because risk factors, causes, and treatments vary according to forms and subtypes (
Figure 31). Drugs used to treat this disease account for around 15% of the global ophthalmic pharmaceutical market
[145][9]. In addition, the annual cost of dry eye management, including direct healthcare costs (medication and doctor visits), impact on patient quality of life, and reduced work productivity, is estimated at 3.84 billion USD in the US alone
[156][23].
Figure 31. The two main enteropathogenic classifications of dry eye.
2.2. Animal Models
Several animal models of dry eye have been established to mimic the different characteristics of these diseases, inducing the clinical manifestations by different pathways (mechanical or surgical approaches, neural pathway blockage, topical eye drops, iatrogenic immune response, desiccating stress, etc.)
[157][24]. In the case of desiccation stress, animals are housed in a controlled environment where humidity, temperature, and airflow are regulated (from a few weeks to several months) to disrupt the immune homeostasis of the ocular surface to induce ocular dryness
[65,158,159][25][26][27]. The most common animals used to establish DED models are mice, rats, and rabbits, whereas the use of dogs and primates is less frequent
[66,160][28][29]. On the one hand, mouse models are extremely attractive models for this type of pathology (low cost and various gene knockout models). However, the anatomical and physiological features of mice still constitute a challenge for ocular tissue dissection and DED drug development, as ocular drug biodistribution studies are less representative of human reality
[157][24]. An important link between drug delivery and the mucin content study was observed, as the corneal and conjunctival epithelium are the primary absorption tissues for these drugs. Membrane-associated mucins may decrease or increase ocular bioavailability depending on the extent of their role as barrier or retention sites. Ocular barrier function in mice is equivalent to that in humans, despite the substitution of MUC16 (MUC for mucins in human) by Muc4 (Muc for mucins in mouse) in mouse corneal epithelial cells and the fact that extracellular domains of Muc4 and Muc16 are shorter than those of MUC4 and MUC16, respectively
[55,65][6][25]. Therefore, the mouse model can provide an interesting model for studying factors on drug pharmacokinetics. On the other hand, the histoarchitectural features of the rabbit lacrimal gland more closely resemble those of the human lacrimal gland compared to murine models. Rabbit eyes also allow better accessibility to the ocular surface, thus facilitating phenotypic observation
[160][29]. Indeed, clinical assessment, commonly used in humans to diagnose and evaluate objective symptoms of DED, has been particularly used in rabbits and dogs because of their large exposed ocular surface
[27,76,161][11][30][31]. There are few methods available to assess the objective signs of dry eye in humans or animals, such as the Schirmer’s test (measuring the aqueous tear production in a given time), tear break-up time (demonstrating tear instability) and fluorescein, lissamine green, or rose Bengal staining (demonstrating ocular surface damage)
[27,76,161][11][30][31]. Nevertheless, standardized procedures for these assessments have not been established for testing new therapies and full diagnosis of dry eye in ophthalmic research using DED animal models. For example, the size of the mouse eyeball does not allow the use of these tests without prior modification
[76,143][5][11]. Additional techniques are also used to further study the ocular surface for clinical diagnosis of dry eye, such as the characterization of the tear film osmolarity, the chemical composition of the tear film, the mucins expression, or the ocular surface inflammation by demonstrating higher expression of conjunctival apoptotic markers
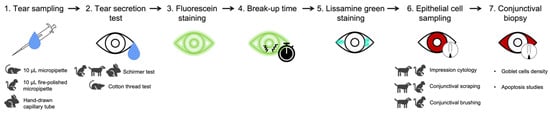
[76][11]. A possible sequence of tests was suggested by Barabino et al. based on commonly used clinical tests and animal models to fully characterize dry eye after highlighting their intrinsic advantages and limitations (
Figure 42)
[76][11]. However, an attempt should be made to translate each clinical test to each animal species to optimize the use of animal models and take advantage of each species in dry eye studies. Certain models are suitable for studying DED pathogenesis, whereas other models are more appropriate for examining the therapeutic effects.
Figure 42. Suggested steps to assess tear film and ocular surface in animal models of dry eye.
Mouse Models
The production of mucins in mouse models and their role on the ocular surface have been recently highlighted
[59][7]. Portal et al. reviewed the different mucin expressions using DED mouse models
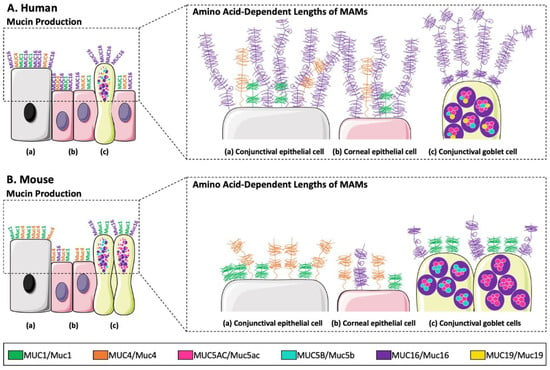
[59][7]. These mucins, conferring the rheological properties of the mucus gel, are similar between humans (MUC) and mice (Muc)
[59,162][7][32]. The three main membrane-associated mucins of the human ocular surface are MUC1, MUC4, and MUC16 (Muc1, Muc4, and Muc16 for mouse). In humans, MUC1, MUC4, and MUC16 are produced in both the cornea and conjunctiva
[163,164][33][34] as well as in the mouse
[165[35][36][37],
166,167], with the exception of Muc16 which is produced only in the conjunctiva
[168][38]. Human lacrimal glands produce mucins MUC1 (also produced by the Meibomian glands), MUC5AC, MUC5B, MUC7, and MUC19, whereas in the mouse, Muc1, Muc2, Muc3, Muc4, Muc5ac, Muc5b, Muc6, Muc10, Muc13, Muc14, Muc15, Muc16, Muc19, and Muc20 are expressed in Meibomian glands
[59][7]. Until now, mucin production by ocular glands in mice has not yet been studied/analyzed at the protein level. The main differences in mucin production between humans and mice are presented in
Figure 53.
Figure 53. Major differences in mucin production by (a) conjunctival epithelial cells, (b) corneal epithelial cells, and (c) conjunctival goblet cells, and in amino acid-dependent lengths of membrane-associated mucins (MUC1/Muc1, MUC4/Muc4, and MUC16/Muc16) between (A) the human and (B) the mouse ocular surface.
Rabbit Models
Various rabbit models have been developed to study DED. A rabbit model was established to simulate Sjögren’s syndrome, a chronic and multisystemic autoimmune disease characterized by lymphocytic infiltration of the exocrine lacrimal glands, leading to the classic manifestations of dry eye (
Figure 31)
[157,161][24][31]. To this end, autoimmune disease can be induced by co-cultivating autologous lacrimal gland cells and peripheral lymphocytes in vitro and injecting them into the lacrimal glands. This procedure allows their dysfunction and induces the symptoms of DED in this model. Other studies have used “short-term” rabbit models for evaporative dry eye by mechanically preventing rabbits from blinking using eyelid specula or stitches
[62,157][10][24]. Due to the use of anesthetics, which may decrease tear secretion, and the induced dry eye acuity, this model is not optimal for studying the pathogenesis of DED, which is a chronic event. However, this model has the advantage of easily and efficiently generating the clinical symptoms of DED in just two hours. Thus, it is very useful for screening and comparing topical eye drops that help maintain cornea hydration, such as artificial tears or other therapies aimed at delaying evaporative loss of the preocular tear film
[124][39]. Moreover, surgical approaches have also been used to develop different DED rabbit models, such as lacrimal or Meibomian gland dysfunction models
[177,178,179,180][40][41][42][43]. Surgical closure of the lacrimal gland excretory ducts or Meibomian gland orifices by cauterization can increase tear evaporation or decrease tear secretion, reflecting a higher electrolyte concentration on the ocular surface. An increase in tear osmolarity on the first postoperative day, accompanied by a significant decrease in conjunctival goblet cell density after 8 weeks, was observed after closure of the lacrimal gland excretory ducts and surgical removal of the nictitating membrane and Harderian gland in rabbits
[76][11]. Moreover, it has been shown that the closure of Meibomian gland orifices is a feature of Meibomian-related dry eye, as seen clinically in several ocular diseases
[180][43]. More recently, a rabbit model in which dry eye is induced by mitogen concanavalin A injection into the orbital lacrimal glands of rabbits has been established
[78,79][44][45]. Concanavalin A induces a strong inflammatory response and destruction of the lacrimal gland structure, creating a clinically relevant situation of acute DED. On the third day after the concanavalin A injection, results showed that tearing was reduced by around half. This model also showed that after a single injection of concanavalin A, induced DED lasts for around a week. However, it is possible to make this model chronic (from days to weeks) for DED by injecting concanavalin A weekly to prolong dry eye symptoms, which are reproducible and consistent for at least 3 weeks. Standard clinical tests for dry eye such as tear break-up time and fluorescein or rose Bengal staining of the ocular surface can be performed much more easily in rabbits due to the large exposed ocular surface and globe size compared to small animals such as mice and rats (
Figure 1 and
Table 1)
[62,76,161][10][11][31]. To have a high chance of acceptable reproducibility under the conditions of Schirmer’s test in rabbits, Barabino et al. suggest a 1-min test without the use of anesthetics
[76][11]. Indeed, it is difficult to compare different conditions used in Schirmer’s test due to the high variability of parameters. For example, the test duration, the use of anesthesia or not, the ease with which the animals are handled, and the opening and blinking of the eyelids can vary the results obtained via Schirmer’s test. Instead of using usual methods such as fluorescein or rose Bengal staining, Goto et al. developed a new tear film stability analysis system using videokeratography
[181][46]. This noninvasive and objective tear break-up time method showed a better sensitivity for tear film stability analysis by capturing consecutive corneal surface images every second, and this technique represents an interesting research area with rabbit models of dry eye due to their cornea size
[76][11]. However, the development of adapted software is required for the specific corneal curvatures of this animal. Therefore, the rabbit model is suitable for the study of lacrimal physiology and pathophysiology of DED, as well as for the efficacity and safety evaluation of therapeutic agents.
3. Ocular Herpes (Herpetic Keratitis)
3.1. Pathology
Herpes simplex virus (HSV) is a widespread viral pathogen that infects most of the world’s population. This contagious infection is transmitted by simple contact with a person carrying the virus or by self-contamination. HSV is a ubiquitous human pathogen represented by two distinct serotypes—HSV-1 and HSV-2—which account for 90% and 20–25% of adult seropositivity, respectively
[182][47]. Many primary infections are asymptomatic, making HSV infections in humans difficult to detect and study. Although oral and genital lesions are the most common manifestations of infection, HSV-1 can also affect ocular tissues, including the cornea, eyelids, conjunctiva, uveal tract, and retina
[183][48]. HSV-1 ocular infection is the leading infectious cause of visual impairment, causing multiple pathologies such as herpes stromal keratitis, which is an immunopathological response caused by recurrent HSV infection of the cornea
[184][49]. This viral form is the most destructive and can lead to blindness due to progressive corneal scarring with recurrences. Herpes stromal keratitis is characterized by progressive leukocytic infiltration, opacity, and vascularization of the cornea
[183,185][48][50]. HSV-1 infection can be classified into primary or recurrent disease. For primary HSV-1 ocular infection, clinical manifestations tend to occur in youth or young adults. After primary infection of the oral-facial region, including the cornea, infected humans are likely to carry a latent viral load because the HSV virus moves particularly to the trigeminal ganglia, where a latent state is established without the production of infectious viral particles
[57,185][50][51]. Subsequently, the virus may undergo cycles of reactivation, causing recurrent viral or immune pathology at the initial site of infection. Thus, frequent attacks of this virus cause nerve damage that reduces the sensitivity of the eye. Much work is being devoted to the HSV-1 study because a thorough understanding of the HSV-1 disease process could lead to the prevention of acute HSV-1 infection, reactivation, development of HSV-1 vaccine, and more effective treatments of recurrent eye diseases in general
[57,185,186][50][51][52]. Clinical trials for vaccines against this type of infection have been ongoing for more than three decades
[186][52]. Despite this, no vaccine has been approved, and no formal clinical trials have evaluated the impact of HSV vaccines on ocular health. Compared with other external anatomic sites, the pathology and healing of corneal tissues after HSV infection is complex and clinically problematic due to the need to preserve corneal clarity and sensitivity, especially in the cases of severe HSV infection, i.e., herpes stromal keratitis
[185,187,188][50][53][54]. This disease is often studied to develop an effective vaccine against HSV-1 because if the vaccine protects against herpes stromal keratitis, it should also protect against other herpes infections in the eye. Therefore, the development of a vaccine is of practical interest against HSV and would confer immunological protection without causing irreversible corneal immunopathology, which is of paramount importance to clinicians and patients.
3.2. Animal Models
Primary and latency HSV-1 corneal infections have been studied in a variety of animal models to better understand multiple aspects of HSV biology, molecular biology, pathogenesis, disease, and immunity, especially for vaccine-induced protection
[57,185,186][50][51][52]. Although all animal models are inherently imperfect representations of human disease, the high species specificity of HSV-1 allows for the development of a wide range of animal models, such as mice, rabbits, guinea pigs, rats, owl monkeys, and rhesus macaques. These models exhibit many characteristics of human HSV-1 corneal disease and are dependent on important experimental parameters, including species, age, and genotype of the animal, the route of infection, as well as the viral serotype, strain, and dose
[57,182,183][47][48][51]. The most popular animal models have been developed in mice and rabbits, followed by guinea pigs, to study ocular HSV-1 latency, reactivation, and recurrence in immune responses and pathogenesis. Biosafety level 2 laboratory facilities with adequate practices and procedures are also required to study HSV and manipulate and house animals. In the case of studies on the mechanisms of HSV infection using animal models, such as the establishment and maintenance of viral latency, these experiments very often require that the animals be kept alive for at least a month in most cases and longer in certain circumstances, resulting in a non-negligible cost
[182][47]. For long-term experiments, a proportion of infected animals will experience significant morbidity and may progress to a fatal outcome, either by acute spread of the virus before the establishment of latency or by reactivation of the latent virus. Furthermore, an important parameter to be considered is the age of the animals because resistance to HSV disease is reduced in young animals whose immune and adaptive response is more vulnerable
[182][47]. Some ocular tissue inoculation approaches are well suited to establish HSV disease as a peripheral infection. Viral inoculum is injected into normal or scarred corneas to facilitate viral uptake and mimic sites of primary human ocular HSV-1 infection. The disease progression is measured by examining corneal opacity and lesions
[182][47]. Using this invasive model, core body temperature, coat appearance, weight, posture, movement capacity, and aggressiveness are measured to assess the animal’s pain and discomfort and visually distinguish moribund animals from those that appear normal. After initial replication in the periphery, HSV-1 infects the ophthalmic branch of the trigeminal nerve and can be detected in the trigeminal ganglia within 2 to 3-days of infection
[189][55].
Mouse Models
Several features make mouse models excellent candidates for studying HSV-1-induced immune responses during latency, reactivation, and recurrent HSV-1 infection. Although murine models of ocular primary infection are intrinsically different from human herpes stromal keratitis, many parallels can be drawn to clarify vaccine efficacy. Murine models are used in basic research to assess the ocular pathogenesis of acute HSV 1 infection and characterize immune responses
[186,191][52][56]. The availability of inbred and transgenic strains for studying this pathology is greater than for other species, and reagents are also available to dissect the immune response to HSV-1
[57,182,183,185][47][48][50][51]. The knockout mouse models used for ocular herpes involved the elimination of components of the immune system and are used to study the effects of the immune system on latency and reactivation. These transgenic mouse models have provided insight into the role of specific genes and cytokines involved in HSV-1 ocular disease, i.e., HSV-1 latency, reactivation, and recurrence
[57,182,183][47][48][51]. Among these models, Human
ApoE3+/+ and Human
ApoE4+/+ knockout mice were developed to study the role of human
ApoE4 [192][57]. This gene plays a role in the establishment of HSV-1 latency, being implicated in the pathogenesis of ocular herpes and the immune response of microglia.
ApoE knockout mice are resistant to the neurovirulence of the HSV-1 strain 17Syn
+ after corneal inoculation of the HSV-1 strain, whereas wild-type mice showed a latent load of the virus in the digestive tract. Other transgenic mouse models have been used to study ocular HSV-1 infection, such as
IL-1ra−/− (role of IL-1 in HSV-1 stromal keratitis)
[193][58],
p19−/− (role of IL-23 in the severity of HSV-1 ocular lesions)
[194][59], wild-type and
p53−/− (role of p43 in HSV-1 replication)
[195][60]. One of the main limitations of the mouse model, particularly for vaccine development, is that the virus does not spontaneously reactivate and is not excreted on the surface of the cornea in mice, unlike in rabbits and humans
[182,183][47][48]. It has been stipulated that the reactivation process in mice is more effectively blocked, notably by CD8
+ T-cells resident in the trigeminal ganglia and/or that mice are less sensitive to stimuli that induce reactivation. However, it is possible to induce HSV-1 reactivation from latency, shedding, and recurrent herpes stromal keratitis in mice, but the protocols involve raising the body temperature to dangerous levels or exposing the cornea to ultraviolet light
[196,197][61][62]. It is difficult to detect HSV-1 infection and HSV-1 DNA because the volume of the mouse tear film is very small
[57,183,184][48][49][51]. The spontaneous shedding rate of HSV-1 DNA in mice is extremely low, and there are no known reports of spontaneous recurrent lesions in immunocompetent mice. In addition, infection of the mouse corneal epithelium with most strains of HSV-1 requires some degree of scarring
[183][48]. HSV-1 reproducibly establishes latency in the mouse model from the earliest stages of acute infection, with the viral genome reaching the neuronal ganglia within the first 24 h of infection
[198][63]. Thus, mice represent an important starting point for assessing the effect of several experimental parameters on the development of HSV disease.
Rabbit Models
Rabbit models also have specific advantages and disadvantages for studying ocular HSV-1. Rabbit strains that can be used for ocular herpes studies include the New Zealand White, Dutch Belted, and other pigmented rabbits
[2,57][51][64]. Most strains of HSV-1 can infect all the previously mentioned rabbits. In addition, the main advantage of rabbit models, which also applies to guinea pig models, lies in their ability to spontaneously produce HSV-1 reactivation from latency and ocular surface shedding, as in humans, which does not appear to occur spontaneously in mice
[57,182,183,199][47][48][51][65]. Spontaneous reactivation of HSV-1 in rabbits includes viral shedding in saliva and tears, as in human
[200][66]. Thus, the HSV-1 infection in rabbits is more representative of human disease than that of mice
[2,57,201][51][64][67]. Latent rabbits with high phenotype reactive strains have a high rate of spontaneous HSV-1 shedding, and their lesions share similar characteristics with human HSV-1 lesions
[202][68]. Because herpes stromal keratitis infection in humans represents recurrent herpetic disease resulting from HSV-1 reactivation from latency in the trigeminal ganglia and shedding at the cornea, the ability to induce its recurrent form is a useful feature of the rabbit model
[183][48]. Because of this unique feature, this model has also been widely used to evaluate the efficacy of HSV vaccines in controlling the recurrent phase of the disease
[186,203][52][69]. Despite the very limited availability of transgenic rabbit models, Chentoufi et al. introduced humanized
HLA-A*0201 transgenic rabbit model for the purpose of developing a vaccine against primary ocular herpes by studying HSV-1 infection
[202][68]. This transgenic rabbit model produces human HLA-specific and restricted T-cell responses for the study of vaccines based on human CD8
+ T-cell epitopes
[202][68]. In this study, the human herpes lipopeptide vaccine formulation contains three pairs of peptide epitopes derived from the sequence of HSV-1 glycoprotein D and protects against ocular HSV-1 infection. This humanized transgenic rabbit model produces HSV-1-specific CD8
+ T-cells and shows a reduction in recurrent HSV-1 disease after induction of latent HSV-1 infection when it is immunized with the vaccine. Thus, humanized animal models are a welcome advancement in this field, as previous glycoprotein D subunit vaccines have shown promising results in protection against HSV-1 and/or HSV-2
[186,191][52][56]. Consequently, it is increasingly interesting and important to focus on the development of a new vaccine against HSV infection through translational research between animal models and humans and combining clinically relevant assessments of corneal pathology with immunologic studies of vaccine efficacy. Moreover, infectious epithelial keratitis generally persists longer in rabbits, which may be helpful for testing the efficacy of anti-herpetic drugs
[183][48]. However, rabbit strains are very expensive and difficult to obtain, and the relative lack of reagents to dissect the immune response is an additional limitation of this model
[57,182,183][47][48][51]. These issues are important drawbacks in attempting to understand an immunopathological process such as herpes stromal keratitis with a genetic contribution from the host.
4. Corneal Repair and Transplantation
4.1. Pathology
In the United States, the eye injury rate is at least one million each year, and approximately 2000 workers undergo a work-related eye injury requiring medical treatment every day
[12,204][1][70]. This is because the anatomical location of the eye makes the cornea vulnerable through continuous exposure to various abrasive forces, such as fingernails or prolonged contact lenses wear, for example, and mechanical, chemical, and thermal injuries as well as viral or bacterial infections
[12,204,205,206,207][1][70][71][72][73]. However, overall, 75% of ocular injuries are due to foreign bodies or abrasive damages, and nearly 25% are due to chemical burns
[204][70], which remain the most serious cause of corneal wounds
[206][72]. In some of these corneal injuries, especially in the presence of a severe injury or when the injuries are untreated or not quickly enough, the consequences can be critical due to the limited regenerative capacity of the human corneal endothelium, requiring corneal transplantation or eye enucleation and leading to permanent visual impairment
[12,204,206][1][70][72]. Indeed, the inability of adult human corneal endothelial cells to re-enter the cell cycle results in endothelial cell loss and decreased cell density due to injury, infection, aging, and/or disease
[208,209][74][75]. Although corneal transplantation has one of the highest success rates in human transplantation, there is an urgent clinical need to improve this research field by finding an alternative to donors using animal models
[12,209,210][1][75][76]. Data collected between 2012 and 2013 showed that only 1 in 70 patients received a corneal transplant, while more than 12 million people were waiting for a corneal transplant during the same period worldwide
[12,211][1][77]. Organ donation is a complex process involving numerous social, ethical, and legal issues. As the number and techniques of corneal transplantation increase, so does the need for donor corneas, contributing to a shortage of supply and demand, particularly in developing countries. In the cornea, fibrotic repair presents unique challenges that affect both the clarity and shape of the cornea. With the increasing popularity of surgical techniques that alter corneal refractive errors, understanding the mechanisms of corneal repair has gained increasing attention
[60][78]. The cornea has unique anatomical, cellular, molecular, and functional characteristics that result in significant mechanistic differences in the repair process compared with what occurs in the skin and other organs. When reconstructing a damaged cornea, the most important characteristics of the cornea to consider are its mechanical strength and transparency
[212][79]. Thus, there is a growing demand for preclinical animal models of corneal endothelial dysfunction to evaluate the safety and efficacy of new therapeutics, but it depends globally on the animal model and method used to create the corneal wound (mechanical, thermal, chemical, etc.)
[209][75]. Herein, the researchers are mainly interested in animal models that might be most suitable for both corneal repairs based on their ability to regenerate endothelial cells (
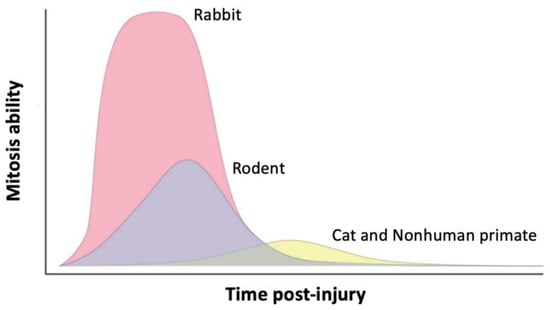
Figure 64) (i.e., the rodents
[60[78][80][81],
61,213], rabbits
[12[1][73][75][76][82][83],
207,209,210,214,215], cats
[109[84][85][86][87][88],
114,115,216,217], nonhuman primates
[217,218,219,220][88][89][90][91] and zebrafish
[208][74]) and for corneal transplantation as host models. In this latter case, the main applications of animal models are the evaluation of basic processes and potential treatments for transplant rejection, as well as the development of innovative approaches to transplantation as alternative solutions to eye bank human eyes, including cell-based therapies and bio-engineered corneal transplants (
Figure 75).
Figure 64. The mitotic capacity of corneal endothelial cells varies depending on the species. The regenerative ability of the human corneal endothelium is consistent with cat and nonhuman primate models. Reproduced from
[209][75]. Copyright (2021), Annals of Translational Medicine.
4.2. Animal Models
Animal Models for Corneal Wounds
The multi-stage process of corneal wound healing is universal for all species, regardless of the nature of the corneal injury
[221][92], and is generally characterized by the enlargement, migration, and proliferation of cells adjacent to the wound edge
[209][75]. However, the mitotic ability of corneal endothelial cells varies considerably between species, which impacts the rate and capacity of endothelial regeneration (
Figure 64)
[209][75]. Rabbits are most frequently used for in vivo research of corneal endothelial cell therapy since they share characteristics with the human corneal endothelium, such as diameter (which allows the use of the same surgical instruments as in humans), repair mechanisms, thickness, and composition
[222][93], but also by their human-like eye size, relatively low cost, and ethical considerations
[78,79,217][44][45][88]. In addition, parameters such as corneal endothelial density, central corneal thickness, and corneal diameter decrease with age in rabbits, as in humans
[222][93]. Nevertheless, the rabbit cornea has corneal endothelial cells that have a high capacity for in vivo regeneration in contrast to the canine, feline, or nonhuman primate models, which have limited corneal endothelial cell proliferation mechanisms like those in human corneas
[207,209,223][73][75][94]. The consequences of this proliferation in rabbits have shown that up to 50% of the central cornea can be repaired within 10 days
[224][95]. This type of result suggests careful attention to the analysis of negative controls to ensure that any endothelial regeneration observed is not the product of native corneal endothelial cell proliferation. The use of rabbits as a model for human corneal healing is limited due to their ability to recover from injury, making it difficult to establish the true efficacy of the treatment tested
[222][93]. Several studies are using nonhuman primate and feline models to overcome these drawbacks
[115,207,209,217][73][75][86][88]. Nevertheless, the use of older rabbits, at least 9 to 12 months of age, may be an appropriate option because they have shown a lower corneal endothelial cell density than younger rabbits
[225,226][96][97]. Going further, Valdez-Garcia et al. showed that 18-month-old New Zealand White rabbits (young adults) are a suitable model for studying human corneal endothelial repair since the mitotic activity of these rabbits decreased significantly with age
[222][93]. This model did not show mitotic activity 72 h after cryoinjury, confirming the delay in corneal endothelial healing in older rabbits. Murine models can also be used for corneal repair
[227,228,229][98][99][100]. To enable us to take advantage of these features, Fini et al. developed a mouse model of penetrating keratectomy (surgical or laser removal of part of the cornea) by adapting a previously successful rabbit model, even though mouse eyes cause problems for surgical manipulation
[60][78]. Another study highlighted that mouse models generated by genetic targeting and/or transgenic techniques are valuable tools to elucidate the role of proteins in the extracellular matrix for corneal wound repair
[61][80]. Finally, an interesting study showed that the zebrafish corneal endothelium can rapidly repopulate on its own and re-enter the cell cycle, after surgical injury, to repair the wound, unlike humans. Thus, the zebrafish model has the potential to regenerate most of its corneal endothelial cells and examine whether the signaling pathways act differently in the injured zebrafish cornea. Indeed, the ease of genetic manipulation of the zebrafish allows for the study of the molecular mechanisms of corneal endothelial regeneration in vivo, which is not possible in other model systems, and understanding the mechanism of cell cycle arrest in human corneal endothelial cells
[208][74].
Figure 75. New approaches of cell-based therapy for corneal endothelial cell transplantation. Adapted from [230]. Copyright (2021), Elsevier, Experimental Eye Research. New approaches of cell-based therapy for corneal endothelial cell transplantation. Adapted from [101]. Copyright (2021), Elsevier, Experimental Eye Research.
Animal Models for Corneal Transplantation
Although humans normally have a sufficient corneal endothelial cell density for a lifetime, they may have excessive corneal endothelial cell loss because of previous eye surgery or some pathologies (e.g., Fuchs endothelial corneal dystrophy
; Section 2.5), posterior polymorphic corneal dystrophy, herpetic viral infections, trauma, or elevated intraocular pressure, which can cause irreversible corneal endothelial dysfunction and decompensation, and thus loss of transparency and corneal blindness
[217][88]. Traditionally, full-thickness corneal transplantation has been the only choice for restoring vision. Nevertheless, other corneal diseases, such as corneal neovascularization
(Section 2.4), can cause graft rejection. Alternative corneal endothelial cell therapies have also attracted considerable research interest to circumvent the routine use of human donor corneas (limited number of donors, lack of quality, and complexity of surgery) or avoid the use of full-thickness corneas. Over the last decade, endothelial transplantation has thus emerged, notably to avoid graft rejection, primary graft rejection, and progressive decrease in cell density
[35,207,231,232][73][102][103][104]. Appropriate models are needed to evaluate the results of cell therapy and determine the safety of the procedure to restore corneal endothelial cell function. Differentiation of pluripotent stem cells and generation of corneal endothelial cells from other cell sources as animals are very promising approaches for the development of cell therapy to treat corneal endothelial disease
[35,207,230][73][101][102]. Endothelial cell transplantation from different human or animal cell sources and recipients as a host animal is possible because of the immune privilege of the anterior chamber, especially in the case of host-incompatible grafts that would otherwise be rejected at other transplantation sites
[35,230][101][102]. Currently, the two main methods investigated to deliver live corneal endothelial cells with sufficient potential to adhere to the posterior cornea are the injection of cells into the anterior chamber of the eye and the implantation of carriers or scaffolds to perform bioengineered corneal endothelial grafts (
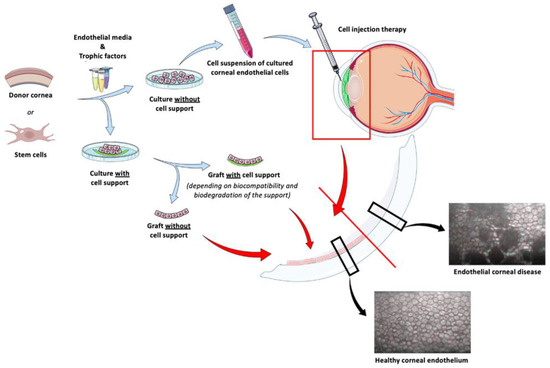
Figure 75)
[207,210,217,230][73][76][88][101]. On the one hand, the cell injection method relies on the simplicity of the technique and could be implemented worldwide, even in regions without access to highly trained corneal experts. On the other hand, the carrier materials (e.g., membranes of amniotic, silk fibroin, collagen I, gelatin, or hydrogels) must meet certain criteria such as biocompatibility, optically transparency, ease of surgical manipulation, and demonstration of mechanical properties like those of the native cornea
[35,207][73][102]. Animal models offer a multi-level approach, integrating macro- and micro-environmental influences, and these models are also necessary to specifically investigate surgical transplantation or implantation of corneal endothelial cells, as well as to investigate issues including biodegradability, immune-tolerance, and long-term outcomes. The most common recipient animal species for in vivo testing are rabbits, followed by rodents, nonhuman primates, and felines, while the most common species used for in vitro corneal endothelial cell culture and transplantation are humans, followed by rabbits, nonhuman primates, felines, and murine
[207,210,217,230][73][76][88][101]. Murine models have provided a better understanding of the pathogenesis of immune rejection and a wealth of information on the immune graft rejection process, thanks to the different rodent strains available
[35,217,233][88][102][105]. In the event of rejection, the graft then becomes opacified due to an immune reaction
[35,233][102][105]. This opacification is the result of immune cell-induced damage to the graft endothelium, leading to edema. A temporary corneal opacification is observed in mice and rats in the days following graft transplantation
[35][102]. Applying clinical criteria for graft rejection, this temporary opacification may be significant enough to be considered complete graft rejection. Thus, the opacification degree and duration are necessary to determine non-reversible rejection. The C57BL/6 and BALB/c mouse strains are the most widely used to investigate the innate immune system and immunological processes during graft rejection. Recently, a study compared several aspects of corneal rejection using these mouse models
[233][105]. The results indicated that M1 macrophages appear to play a crucial role in the rejection process. Furthermore, the authors suggest that the BALB/c recipient model could be used as a surgical control for corneal transplantation experiments, while models using C57BL/6 as recipients can serve as transplantation models in a clinical context considered “high-risk” due to severe inflammation and a high rejection rate. Strain-dependent differences then convey different innate immune responses in BALB/c and C57BL/6 strains, suggesting the mouse lineage of donor and recipient animals must be carefully considered. However, from a clinical point of view, mouse and rat models show significant anatomical differences from human grafts
[207,210,217,230][73][76][88][101]. It also remains unclear to what extent these models mimic the immunological mechanisms of corneal graft rejection in humans, and whether differences in the innate immune systems of these two mouse strains affect outcome after corneal transplantation
[35,233][102][105]. Consequently, the dissimilarity between murine and human immune systems, as well as the inherent size difference between the species, have made the use of larger animal models essential. Indeed, larger animal models facilitate surgery and allow the use of clinical techniques familiar to the ophthalmologist to assess graft rejection and study changes in the endothelium
[35,207][73][102]. In addition, the selection of the animal model that most closely resembles human anatomy and physiology is desired to facilitate the transfer of developments from the animal model to humans when creating the corneal endothelium, especially for carrier implantation
[35,207,230][73][101][102]. In contrast to murine models, the use of larger animal models, such as rabbits, is primarily intended for the study of graft rejection, enabling corneal transplants to be performed that will be more easily transferable to human transplantation due to their similar eye size between large species
(Figure 1 and Table 1). The rabbit shows various signs of rejection also seen in humans, such as retro-corneal membranes, epithelial decompensation, and neovascularization of the graft
[35][102]. It has also been shown in rabbits that graft size and location contributed to rejection. Indeed, widespread rejection was observed for 7-mm grafts
[234][106], whereas smaller grafts, 4 to 5 mm, did not appear to induce rejection since the grafts retained their transparency
[235][107]. Also, grafts placed closer to the limbus have higher rejection rates, particularly in the case of vascularization
[235][107]. Finally, although the ideal research model for human application is the nonhuman primate for obvious reasons, in 2016, Bostan et al. showed in vivo functionality of a corneal endothelium transplanted by cell injection therapy in a feline model as an intermediary model and were able to restore corneal clarity and thickness up to 7 days post-transplant
[115][86]. To date, the different animal models have proven to be complementary in providing researchers and clinicians with a means to develop new surgical techniques as well as to evaluate the function of various corneal grafts and new cell therapies.
5. Corneal Neovascularization
5.1. Pathology
The avascular structure of the cornea (no lymphatic or blood vessels) is one of the main reasons for the optical clarity of the cornea and appears to be an “angiogenic privilege” against corneal infiltration by blood vessels
[236,237][108][109]. Homeostasis exists in the cornea, where pro-angiogenic and anti-angiogenic factors are in equilibrium. Up-regulation of proangiogenic factors accompanied by down-regulation of antiangiogenic factors prompts the formation of new blood vessels in the avascular corneal stroma, called corneal neovascularization. Thus, hypoxia or inflammation (secondary to infection, trauma, or graft rejection) triggers the production of growth factors and angiogenic signals in response to tissue aggression
[236,238,239,240][108][110][111][112]. These new vessels, initially immature and poorly structured, diffract light and introduce proteins, lipids, and inflammatory cells that disrupt corneal immune privilege, promote further inflammation, prompt graft rejection and corneal scarring.
5.2. Animal Models
Overall, the main models for corneal neovascularization are from rabbits, rats, and mice for this type of pathology
[1,238,240,241,242][110][112][113][114][115]. Many different methods have been used to induce corneal neovascularization in animals. Among them, alkali burn and suture placement are the two most widely used models for studying mechanisms and developing therapies
[240,243,244,245,246][112][116][117][118][119]. In the alkali-induced model, corneal neovascularization can be induced by placing NaOH-soaked paper on the ocular surface of the animal for 10 s. In the suture-induced model, corneal neovascularization can be triggered by suturing two 10-0 nylon stitches directly onto the cornea
[245][118]. In both models, corneal neovascularization appears and progressively extends over the first two weeks following induction of neovascularization in the animal, with up-regulation of vascular endothelial growth factors (VEGF). A strategy for treating corneal neovascularization is to inhibit VEGF activity by competitively binding it to an anti-VEGF antibody
[240,244,245,246][112][117][118][119]. VEGF is one of the main factors involved in the pathogenesis of corneal neovascularization. The use of animal models is, therefore, essential to establish safe doses and administration methods before these agents can be used in the clinical context and justify further development of these agents. The efficacy of anti-VEGF agents depends on how quickly treatment is initiated after the onset of corneal neovascularization. Early administration of treatment on day 1 post injury inhibits corneal neovascularization more effectively in an experimental rabbit model of limbal insufficiency than when the treatment is administered on day 14 post injury
[247][120]. However, treatment of corneal neovascularization with the anti-VEGF antibody has certain limitations. It is only a symptomatic treatment for corneal neovascularization and does not cure the cause of the disorder. In some cases, repeated treatment is required to maintain the drug’s positive effect over a period of time
[244][117]. In addition, the affinity of anti-VEGF agents for VEGF needs to be considered in animal models since it may be lower than in humans, as is the case in rat models
[244][117]. Consequently, further research in animal models is required before anti-VEGF agents can become key therapeutic agents in the inhibition of corneal angiogenesis. Some studies have shown that the use of gold or silver nanoparticles can improve therapeutic treatments of corneal neovascularization in murine models by increasing drug residence time and targeting
[248,249,250][121][122][123]. In a study, these nanoparticles functionalized with a heparin derivative demonstrated efficacy as anti-angiogenesis agents
[248][121]. Moreover, the use of gold nanoparticles, topically applied, significantly reduced the development of corneal neovascularization induced by alkali burn, without any significant side effects, by inhibiting the extracellular signal-regulated kinase pathway
[249][122].
6. Corneal Dystrophy
6.1. Pathology
Corneal dystrophies represent a heterogeneous group of genetic diseases generally describing rare inherited disorders of the cornea that are bilateral and often symmetrical, slowly progressive, and unrelated to environmental or systemic factors
[251,252,253,254][124][125][126][127]. They are characterized by abnormal accumulations of insoluble deposits in different layers of the cornea and affect cells, tissues, and/or organs. However, there are many exceptions, as not all corneal dystrophies meet these criteria
[251][124]. In 2015, the International Committee for the Classification of Corneal Dystrophies (IC3D) revised the anatomic classification of corneal dystrophies (epithelial and subepithelial, epithelial-stromal TGFBI, stromal, and endothelial), and this classification also identifies corneal dystrophies into fcategories based on clinical, pathologic, and genetic information
[251,252,253][124][125][126]. Moreover, many dystrophies involve more than one corneal layer. A total of 22 distinct forms of corneal dystrophies that are inherited through autosomal dominant patterns can be distinguished, although autosomal recessive and X-chromosomal dominant patterns also exist
[251,254][124][127]. The symptoms of patients with corneal dystrophy are highly variable
[252][125]. Many of them do not show any symptoms, especially at the beginning of the disease. Patients usually have recurrent epithelial erosions resulting in morning eye pain and discomfort for those in whom the more superficial layers are affected
[251,252,254][124][125][127]. In contrast, patients with stromal dystrophies tend to have reduced visual acuity due to deposits of abnormal substances in the main area of the cornea. Vision loss is also the primary symptom in patients with endothelial corneal dystrophies due to fluid retention (swelling of the cornea), leading to corneal edema which results in progressive loss of corneal transparency
[252,254,255,256,257,258][125][127][128][129][130][131]. This type of corneal dystrophy accounts for approximately 60% of all types of corneal dystrophies
[252][125]. The discovery of the genetic basis of corneal dystrophies is not complete, and the molecular mechanisms of the different mutations in the pathogenesis of each corneal dystrophy remain unclear. The development of gene therapy in the initial stages of corneal dystrophies is an important scientific challenge for the future. In contrast to retinal dystrophies, corneal dystrophies are more amenable to such therapy because of the anatomical accessibility of the cornea
[254][127].
Fuchs endothelial corneal dystrophy (FECD) is the most common corneal dystrophy, with a prevalence ranging from 3 to 11% depending on the age, ethnicity, and sex of the population
[255,256,257,258,259][128][129][130][131][132]. This genetically heterogeneous disease is the most frequent cause of corneal transplantation worldwide. Two forms of FECD exist—the rare early-onset form and the more common late-onset form
[252,255,257][125][128][130]. Although the primary cause of this disease is unknown
[254[127][133],
260], this bilateral disease of the corneal endothelium is characterized by accelerated loss of corneal endothelial cells and the formation of extracellular matrix excrescences in Descemet’s membrane, called guttae
[256,257,258,259][129][130][131][132]. Endothelial cell oxidative stress, apoptosis, loss of pump function, and deposition of abnormal extracellular matrix occur in the initial stages of the disease. These responses are manifested by endothelial cell loss, enlargement, and change in morphology associated with Descemet’s membrane thickening and guttae formation, leading to corneal edema until vision is lost. Although FECD is primarily a disease of the corneal endothelium, secondary changes may eventually affect all layers of the cornea, such as the stromal and epithelial layers, as well as the corneal nerves
[255,258][128][131]. It is inherited in an autosomal dominant mode, with incomplete penetrance and a female predominance
[255,257,258][128][130][131]. Other corneal diseases described in this review could be associated with FECD, such as diabetes mellitus (DBMT
; Section 2.6) and keratoconus
(Section 2.7) [251,257][124][130].
6.2. Animal Models for Fuchs Endothelial Corneal Dystrophy
Currently, there is no treatment for FECD other than corneal transplantation
[259][132]. Descemet’s membrane stripping is a major step in endothelial keratoplasty techniques for this disease in the context of corneal transplantation. Preclinical animal models of Descemet’s membrane stripping have been used to evaluate various biological and synthetic support materials seeded with cultured corneal endothelial cells
[209][75]. A cellular approach has been used to replace corneal endothelium and involves delivering cultured human corneal endothelial cells as a cell suspension directly into the anterior chamber by intracameral injection after removing native corneal endothelial cells by stripping the Descemet’s membrane
[255,257,260,261][128][130][133][134]. Okumura’s group demonstrated that Descemet’s membrane stripping, in combination with cultured corneal endothelial cells injection, is feasible for FECD patients to further improve visual quality using a rabbit and monkey model
[220,262][91][135]. The application of a Rho-kinase (ROCK) inhibitor Y-27632, after injection of cultured corneal endothelial cells in monkey and rabbit models, has been shown to improve cell adhesion and proliferation by regenerating healthy corneal endothelium
[262,263][135][136]. This method also restored vision and maintained corneal transparency without any drug side effects, such as persistent epithelial defects or corneal stromal scarring. Researchers have achieved similar results by implanting human corneal endothelial cells in primates
[220][91]. Y-27632 also effectively reduces FECD-induced central corneal edema in a small group of patients
[264][137] and improves wound healing in rabbits and primates
[264][137]. A study also showed that after culturing normal corneal endothelial cells and FECD cells to create corneal endothelium, the modified FECD corneas were transplanted onto devitalized human stromal media into feline eyes and were able to restore corneal clarity, observed with a slit-lamp biomicroscope, and corneal thickness up to 7 days post-transplant in these animals
[115][86]. These tissue engineering models demonstrate that cell therapy with human corneal endothelial cells delivered as a cell suspension to the anterior chamber can produce in vivo functional corneal endothelium and is well tolerated
[257][130]. They also make it possible to study the behavior of FECD cells in a healthy environment as well as to analyze and understand the initial events of this disease. In addition, injected corneal endothelial cells in the context of corneal transplantation represent an important source of FECD cells to study the late cellular events of the disease
[258][131].
Animal models allow for the study of the genetics of FECD at different stages of the disease and/or in the presence of external stress factors, as well as for the testing of new treatments for FECD
[209,256,257,258,260][75][129][130][131][133]. The genetic basis of FECD includes many genes and chromosomal loci, although alterations in the
TCF4 gene are responsible for approximately 70% of FECD cases
[209,255][75][128]. Although mutations causing early-onset FECD have been exclusively linked to the α2 chain of collagen VIII (COL8A2), which is a major component of Descemet’s membrane, the
TCF4,
TCF8,
SLC4A11,
ZEB1,
LOXHD1 genes have been implicated in late-onset FECD
[209,261][75][134]. A homozygous double knockout mouse model for
COL8A1 and
COL8A2 was generated
[265][138]. The authors observed anterior segment dysgenesis of the eye and anterior chamber protrusion was observed, as well as thinning of the corneal stroma and Descemet’s membrane. However, no guttae were observed, and there was no evidence of corneal opacification. Homozygous knock-in mouse models containing a point mutation homologous to the human Q455K and L450W mutation in the
COL8A2 gene were also generated as these missense mutations have been shown to cause early-onset FECD in humans
[266,267][139][140]. These knock-in mice showed endothelial phenotypes like human FECD at early onset, including altered corneal endothelial cell morphology, their loss, guttae formation, endoplasmic reticulum stress, and activation of the corneal endothelial unfolded protein response
[209,257,258][75][130][131]. Induction of autophagy by lithium administration and
N-acetylcysteine ingestion via drinking water reduce both endoplasmic reticulum and oxidative stress and increase corneal endothelial cell survival in Q455K and L450W mouse models
[268,269][141][142]. Taken together, these results support a pathogenic mechanism in which early-onset FECD is the result of endoplasmic reticulum stress and unfolded protein response-associated apoptosis rather than a loss of function of
COL8A2. To investigate the role of various genes (
TCF4,
SLC4A1, and
ZEB1) involved in late-onset FECD, genetically engineered mice were also generated
[209,257][75][130]. Mouse models have been used to study the
TCF4 gene
[257][130]. Although the
TCF4 heterozygous mouse models are viable,
TCF4 homozygous knockout (
TCF4−/−) mice die within 24 h of birth, indicating that
TCF4 is a crucial transcription factor required for normal development
[270][143]. In addition,
TCF4−/− mice do not have any anatomical defects, including no specific ocular abnormalities, which limits the ability to study corneal endothelium
[271][144]. However,
TCF4−/− mice used to study FECD showed disturbed hindbrain development that is not characteristic of FECD
[257][130]. In addition, researchers have focused on the
SLC4A11 gene that encodes NaBC1
[257,272][130][145]. Mutations in the
SLC4A11 gene have been shown to cause either congenital hereditary endothelial dystrophy or combined hearing and vision impairment
[272][145]. Therefore, a homozygous mutant
SLC4A11 knockout mouse model
[273][146] was developed to have a mild corneal phenotype, with no significant difference in corneal endothelial morphology and without any opacification or edema but significant abnormalities of the audio-vestibular system. However, the main phenotypic change observed in the cornea was an increase in the absolute and relative height of the basal corneal epithelial cells
[273][146]. The lack of a severe phenotype in the cornea could be due to compensatory mechanisms in the knockout mouse or because NaBC1 does not play a direct role in maintaining corneal clarity in the mouse, despite its strong correlation with FECD and congenital hereditary endothelial dystrophy in humans. Nevertheless, understanding why mouse corneas do not exhibit a dystrophic phenotype may provide insight into the molecular basis of FECD, as well as conditions such as congenital hereditary endothelial dystrophy. Homozygous and heterozygous
ZEB1 knock-out mutant mice were generated that exhibit ectopic expression of epithelial genes in the corneal endothelium and keratocytes. These mice also exhibit more features of posterior polymorphic corneal dystrophy, rather than that seen in FECD
[274][147]. Therefore, the development of effective genetic interventions to treat corneal dystrophies is hampered by the lack of animal models resembling human corneal dystrophies, making it difficult to evaluate in vivo treatments.
Another approach is being considered to develop new animal models of FECD. Corneal endothelial cells are particularly sensitive to oxidative stress, which is the case in FECD patients due to their chronic exposure to UV radiation and the high oxygen demand associated with active pump function
[209][75]. In general, the severity of UV-induced tissue damage depends on the wavelength and intensity of the light and the absorption spectrum of each tissue. In addition, UV irradiation leads to a slow onset of corneal endothelial cell damage
[209][75]. The effect of UVA has been shown to be an important etiologic factor in FECD pathogenesis, explaining the predominantly central location of cell loss and guttae formation in FECD patients
[257,275][130][148]. Gene expression studies of corneal endothelial cells from human FECD patients and an FECD mouse model provide evidence of accelerated senescence as a potential consequence of oxidative stress
[258][131]. To mimic a pro-oxidative environment leading to DNA damage and resulting corneal endothelial dysfunction, UV irradiation was applied to animal corneas
[209,257,258][75][130][131]. At the molecular level, UVA exposure induced delayed nuclear DNA damage with low corneal endothelial cell density in mice starting only one month after irradiation
[257][130]. UVB irradiation induced alterations in corneal endothelial cells in mice, rats, and rabbits, with corneal endothelial cells apoptosis and corneal edema
[209][75]. Liu et al. recently developed a nongenetic UVA-induced FECD mouse model
[275][148]. The corneal UVA exposure time was adjusted to obtain the appropriate fluence, i.e., 250 J/cm
2, 500 J/cm
2, 750 J/cm
2, and 1000 J/cm
2. To simulate the life-long exposure of endothelium to UV light, the authors used high-dose UVA irradiation (1000 J/cm
2) and detected progressive degenerative effects of UVA-mediated damage. This in vivo model characterizes FECD in patients as morphologic changes, corneal endothelial cells loss, Descemet’s membrane thickening, and guttae-like lesion formation
[209,257,258,275][75][130][131][148]. This late-onset mouse model, based on the corneal endothelial cell sensitivity to oxidative stress, simulates the female predisposition as observed in FECD patients and showed more pronounced cell loss and corneal edema in female mice at a lower dose of UVA compared with male mice
[275][148]. This method may be the most physiologically relevant inducible animal model of FECD currently under development and provides a tool to study potential therapeutic interventions for all forms of FECD, regardless of genotype. In the same idea, the study of corneal endothelial dystrophy in a canine model suggests the underlying presence of an inherited component by sharing clinical and histologic similarities with corneal endothelial dystrophy in human patients
[209][75]. However, it is important to note that preclinical studies in laboratory animals are often poor predictors of human clinical trials. This is due to the strictly regulated breeding environments, their highly inbred and uniform genetic backgrounds, and the lack of accounting for environmental factors associated with the risk of developing corneal endothelial dystrophy, such as smoking, diabetes, and cardiovascular disease. To improve the relevance of these animal models, it is crucial to consider and expose them to the same epigenetic factors as their human counterparts
[209][75].