Addressing the global environmental problem of water splitting to produce hydrogen fuel by solar energy is receiving so much attention. In water splitting, the essential problem to solve is the development of efficient catalysts for oxygen production. In the generation of hydrogen by water splitting, molecular oxygen should be evolved at the same time. However, it is not so easy to artificially achieve photosynthetic oxygen generation by developing novel systems in a short period of time, which took one billion years in the natural world. The oxidation of water to produce oxygen, i.e., oxygen evolution reaction (OER), involves transfers of four electrons and four protons, while the reduction of water to produce hydrogen, i.e., hydrogen evolution reaction (HER), is a reaction of two electrons and two protons.
- oxygen evolution reaction
- catalysis
- mechanism
- artificial photosynthesis
- titanium oxide
- bismuth vanadate
- metal oxides
- Fourier transform infrared spectroscopy
- density functional theory
- metal oxide
1. TiO2
2. BiVO4
3. SrTiO3
4. Ga2O3
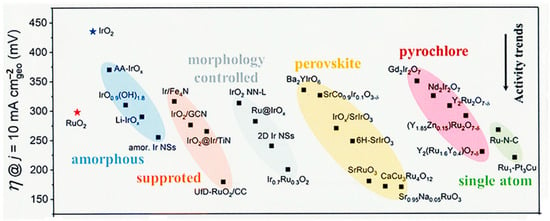
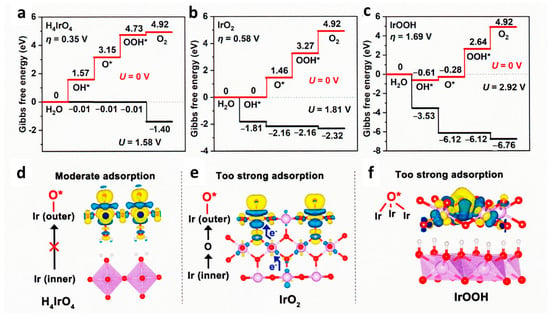
5. IrO2 and RuO2
6. Perovskite as Electrocatalysts
7. Transition Metal (TM) Compounds
Multicomponent transition metal oxides and (oxy)hydroxides are the most promising OER catalysts due to their low cost, adjustable structure, high electrocatalytic activity, and outstanding durability. Co-, Ni-, and Fe-based OER catalysts have been considered to be potential candidates to replace noble metals, especially for electrocatalysts, due to their tunable 3d electron configuration and spin state, versatility in terms of crystal and electronic structures, as well as abundance in nature [67]. The latest advances in the rational design of the related OER electrocatalysts and the modulation of the electronic structure of active sites were comprehensively summarized, besides a brief overview of the mechanisms of OER and the theory and calculation criteria [68]. Wang et al. reviewed the fundamental understanding of the electronic structure of low-cost TM oxide-based catalysts for electrochemical OER, and its relationship with the catalytic activity and the reaction mechanism was discussed [69]. Feng et al. reviewed the relationship between TMs and OER catalyst activity, and then the mechanism of synthesis strategy in different types of TMs-based catalysts was summarized [70]. Guo et al. reviewed the state-of-the-art amorphous transition metal-based OER electrocatalysts, involving oxides, hydroxides, sulfides, phosphides, borides, and their composites, and then the practical application and theoretical modeling of the OER mechanisms in the OER were presented [71]. Though transition metal phosphides often exhibit excellent HER activity, the OER catalytic performance is not outstanding. Huang et al. reviewed the strategies for preparing highly active OER catalysts of transition metal phosphides [72]. The early transition metals (Ti, V, and Cr) can form very stable M=O units, while the late transition metals (Ni and Cu) can only theoretically form unstable M=O structures. On the other hand, for Mn, Fe, and Co, the metal-oxo motif switches between two valence tautomers in the form of Mn+1 = O2− and Mn–O•−. The former with an electrophilic oxygen atom can proceed via the acid–base WNA pathway to form the O–O bond, whereas the latter favors the oxygen radical coupling pathway for O–O bond formation [73].7.1. CoOx
CoOOH was selected as the OER co-catalyst of aluminum-doped strontium titanate (SrTiO:Al) photocatalyst to attain almost unity in the internal quantum efficiency of UV-induced water splitting with Rh/Cr HER co-catalyst [74]. The recent progress of Co3O4-based electrocatalytic materials for the acidic OER was presented with particular reference to the catalytic mechanism and guidelines for the design principles from both experimental and theoretical perspectives [75]. Afterward, emerging strategies were outlined to improve the catalytic performance of Co3O4-based acidic OER catalysts, including phase engineering, component regulation with doping, composite with carbon-based materials, and multi-phase hybridization [75]. For the application of Co oxides to photocatalysts, operando XPS measurements were performed. The catalyst undergoes chemical–structural transformations as a function of the applied anodic potential, with complete conversion of the Co(OH)2 and partial conversion of the spinel Co3O4 phases to cobalt oxyhydroxide, CoO(OH), under precatalytic electrochemical conditions. This interpretation revealed that the presence of Co(OH)2 enhances catalytic activity by promoting transformations to CoO(OH) [76]. To study the mechanism of OER on CoO(OH), operando X-ray absorption and Raman spectroscopy revealed that a Co(IV) species, CoO2, is the dominating resting state of the catalyst. Oxygen isotope exchange experiments showed that a cobalt superoxide species is an active intermediate in the OER. This intermediate is formed concurrently with the oxidation of CoO(OH) to CoO2. Combining spectroscopic and electrokinetic data, the rate-determining step of the OER was identified as the release of dioxygen from the superoxide intermediate [77]. By using a water-in-salt electrolyte, the water activity was systematically tuned and the mechanism as a function of applied potentials in water electrolysis was probed. The mechanism is sensitive to the applied potential. The Co-OO-Co bond forms via an intramolecular oxygen coupling mechanism at low potentials, whereas it proceeds through a WNA mechanism by forming Co-OOH at high potentials [78]. The morphology-dependent analysis for well-defined crystalline CoO(OH) revealed that the active sites are exclusively located at lateral facets rather than basal facets. Theoretical calculations show that the coordinately unsaturated Co sites of lateral facets upshift the O 2p-band center closer to the Fermi level, thereby enhancing the covalency of Co-O bonds to yield the reactivity [79]. The sequential oxidation kinetics with Co3O4 nanoparticles involving multi-active sites for water oxidation in the OER catalytic cycle were resolved by applying quasi-operando transient absorption spectroscopy to a typical photosensitization with Ru-dye and sacrificial electron donor. The CoIV intermediate distribution plays a determining role in OER activity and results in the slow overall OER kinetics [80]. The redox process between CoIII and CoIV species does not follow a proton-coupled electron transfer mechanism that is thought to be common prior to the OER, but it involves a proton-decoupled electron transfer, clarified by isotope labeling experiments and in situ electrostatic modulation [81]. An oxygen vacancy (Vo)-rich environment facilitates the reconstruction of Co3O4 to the Co(OH)2 intermediate with proton vacancies (CoIIOx(OH)y), which is favorable for the formation of the active species of CoO(OH). Correlative operando Raman spectra characterizations and electrokinetic analyses indicated that a moderate Vo density can switch the O–O bond formation pathway, from a WNA to an INA pathway, which is more kinetically favorable for water oxidation [82]. As shown in Figure 8b with O vacancy, at step 3, three protons and one electron are removed to form Co-O(OH). At step 4, CoIII sites of Co-O(OH) are oxidized to CoIV which can be deprotonated (step 8) by hole attack oxo ligand CoIV = O forms a Co-O-O triangle (step 9), and then becomes CoII-OO• (step 10). At the next oxidation (step 10), O2 is released and CoII back to CoIII with the coordination of water. The I2M process was excluded based on the experimental results of using the H218O isotope [82].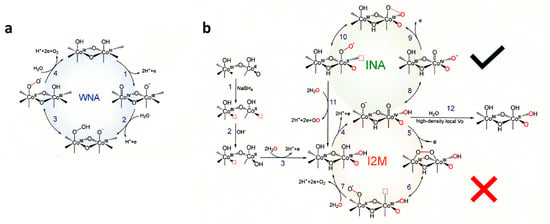
7.2. NiOx
For the nascent ultra-small nickel oxyhydroxide (NiOOH) particles (<3 nm), the thermodynamics of Ni dissolution was calculated by using first-principles theory at a near-neutral pH range, and the mechanism of OER on the γ-NiOOH surface was clarified. It was concluded that (i) ∼4% Ni cations on the surface of γ-NiOOH dissolve at pH = 7 and 1.73 V vs. RHE; (ii) on the pristine γ-NiOOH surface, OER proceeds via the “lattice peroxide” mechanism (*H2O → *OH → *O–OlattH* → O–Olatt → O2) with an overpotential of 0.70 V; (iii) in the presence of Ni cationic vacancies, OER proceeds via the “hydroperoxide” mechanism (*OH + *H2O → *2OH → *OOH → O2) with an overpotential of 0.40 V [85]. For NiOOH-based materials, light-triggered reversible geometric conversion between octahedron (NiO6) and square planar (NiO4) was proposed. The unit cell was undergone to achieve electronic states with alternative metal and oxygen characters throughout the oxygen evolution process. Utilizing this electron transfer pathway can bypass the potential limiting steps, that is, O–O bonding in the AEM and deprotonation in the LOM. As a result, the electrocatalysts that operate through this route showed superior activity compared with previously reported electrocatalysts [86][87]. By incorporating Fe and V into Ni(OH)2 lattices, OER activity was improved. X-ray photoelectron/absorption spectroscopies revealed the synergistic interaction between Fe/V dopants and Ni in the host matrix, which subtly modulates local coordination environments and electronic structures of the Fe/V/Ni cations. Further, in situ XAS analyses manifested contraction of metal–oxygen bond lengths in the activated catalyst, with a short V–O bond distance. DFT calculations indicated that the V site of the Fe/V co-doped NiOOH gave near-optimal binding energies of OER intermediates and had lower overpotential compared with Ni and Fe sites [88]. A series of Mn-, Co-, Fe-, and Zn-doped nickel oxides were investigated by using operando UV−vis spectroscopy coupled with time-resolved stepped potential spectroelectrochemistry. The Ni2+/Ni3+ redox peak potential was found to shift anodically from Mn- < Co- < Fe- < Zn-doped samples, suggesting a decrease in oxygen binding energetics from Mn- to Zn-doped samples. The OER kinetics had a second-order dependence on the density of these oxidized species, suggesting a chemical rate-determining step involving the coupling of two oxo species. The intrinsic turnover frequency per oxidized species exhibits a volcano trend with the binding energy of oxygen on the Ni site, having a maximum activity for the Fe-doped sample as shown in Figure 9. For Ni centers that bind oxygen too strongly (Mn- and Co-doped oxides), OER kinetics is limited by O–O coupling and oxygen desorption, while for Ni centers that bind oxygen too weakly (Zn-doped oxides), OER kinetics is limited by the formation of oxo groups [89].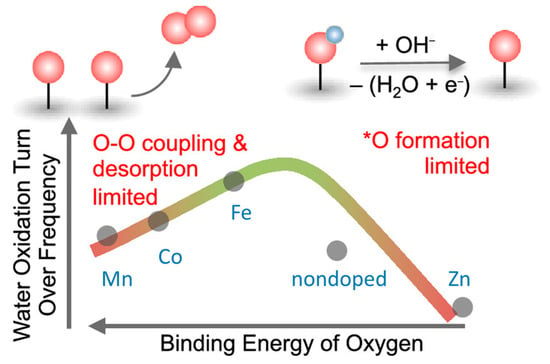
7.3. FeOx
Recent advancement and progress in initializing Fe-based OER electrocatalysts with different supporting materials, including carbon-based materials, layered double hydroxides, Prussian blue analogous, metal–organic frameworks, were reviewed by Xiong et al. [93]. In the review, the OER mechanism and some typical OER electrochemical parameters of Fe-based electrocatalysts supported on various supporting materials from experimental and theoretical viewpoints were highlighted. Some challenges and expectations for promoting the catalytic performance were described [93]. In photoelectrochemical (PEC) water oxidation on hematite (α-Fe2O3), the mechanism of the subsequent rate-limiting O–O bond formation step was investigated by rate law analysis based on EIS measurements and probing the reaction intermediates with operando FTIR spectroscopy. Distinct reaction orders of ~1 and ~2 were observed in near-neutral and highly alkaline environments, respectively. The unity rate law in near-neutral pH regions suggests a mechanism of WNA to –Fe=O to form the O–O bond. Operando observation of a surface superoxide species by FTIR further confirmed this pathway. In highly alkaline regions, coupling of adjacent surface trapped holes (I2M) becomes the dominant mechanism. While both are operable at intermediate pHs, the mechanism switch from I2M to WNA induced by local pH decrease was observed at a high photocurrent level as shown in Figure 10 [94]. In the recent report, transient photocurrent measurements for hematite photoanodes revealed that the OER rate has a third-order dependence on the surface hole density. A mechanism wherein the reaction proceeds by accumulating oxidizing equivalents through a sequence of one-electron oxidations of surface hydroxy groups was proposed. The key O–O bond formation step occurs by the dissociative chemisorption of a hydroxide ion involving three oxyl sites [95].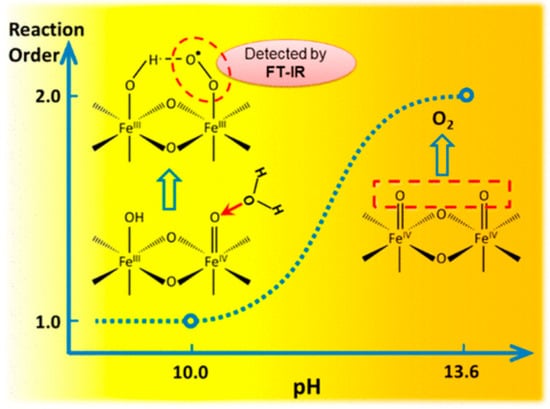
7.4. MnOx
Nature uses a Mn cluster for water oxidation in PS II, and thus, water oxidation using Mn clusters is interesting in artificial water-splitting systems. An ultra-thin manganese oxide (MnOX) was selected as a co-catalyst to modify the surface of BiVO4 photoanode by a spray pyrolysis method [99]. The PEC measurements demonstrated that the surface charge transport efficiency strikingly increased by the MnOX modification. After applying Ar plasma to the BiVO4/MnOx sample, the transport efficiency further increased, and it was around 7 times higher compared with that of pristine BiVO4 samples. The remarkable PEC performance could be attributed to the increased charge carrier density, extended carrier lifetime, and additional exposed Mn active sites on the BiVO4 surface [99]. An α-Mn2O3/FTO electrocatalyst was used in nonaqueous (CH3CN and DMF) and aqueous 0.1 M KPi (pH 7.0) solutions for kinetic studies of heterogeneous water oxidation. The rate of water oxidation was first order in catalyst concentration and in H2O concentration. The square wave and cyclic voltammetry measurements revealed the stepwise proton-coupled electron transfer oxidations of the active MnII–OH2 site to MnIII–OH and then to MnIV=O and finally an electron transfer oxidation of MnIV=O to MnV=O species. The MnV=O species undergoes a rate-limiting O atom transfer to H2O to give a MnIII–OOH2 species that, in turn, undergoes further oxidations to release O2 [100]. An Mn–K cluster was investigated for electrochemical water oxidation. By using XAS, SEM, TEM, XRD, FTIR spectroscopy, and electrochemical methods, it was revealed that conversion into nanosized Mn oxides occurred for the cluster, and the nanosized Mn oxides are the true catalyst for water oxidation [101]. The Mn3O4 nanocatalyst, which exhibits superb catalytic activity for water oxidation under neutral conditions, was analyzed for the complex capacitance. By the change in Mn valence between MnII and MnIV, the charge was accumulated on the catalyst surface prior to the rate-determining O–O bond-forming step. The dissipation ratio was proposed for understanding the energy balance between charge accumulation and charge consumption for chemical O–O bond formation [102]. In Mn3O4 nanoparticles, a profile imaging technique was exploited to understand the correlation between surface atomic structures and the OER. The surface structures of Mn3O4 nanoparticles were changed by the reaction and the surface Mn ions were reconstructed. The commonly considered active sites disappeared from the reconstructed planes, whereas Mn ions were still exposed at the edges of nanoparticles. Thus, the surface reconstructions can deactivate low-index surfaces of Mn oxides in the OER process, which was further validated by DFT calculations [103]. An MnVII=O intermediate during electrocatalytic water oxidation by a c-disordered δ-MnOx was identified as an onset-potential-dependent reduction peak at 0.93 V. This intermediate is proven to be highly reactive and much more oxidative than permanganate ion. Thus, a new catalytic mechanism for water oxidation catalyzed by Mn oxides was proposed with the involvement of the MnVII=O intermediate in a resting state and the MnIV-O-MnVII=O as a real active species for O-O bond formation. Figure 11 shows the proposed catalytic cycle, involving MnVII=O, in the MnOx-catalyzed water-oxidation reaction. The overall mechanistic process involves charge accumulation (S0/S3), charge rearrangement (S3/S4), active-state formation (S4/S4′), and oxygen evolution (S4′/S0) [104].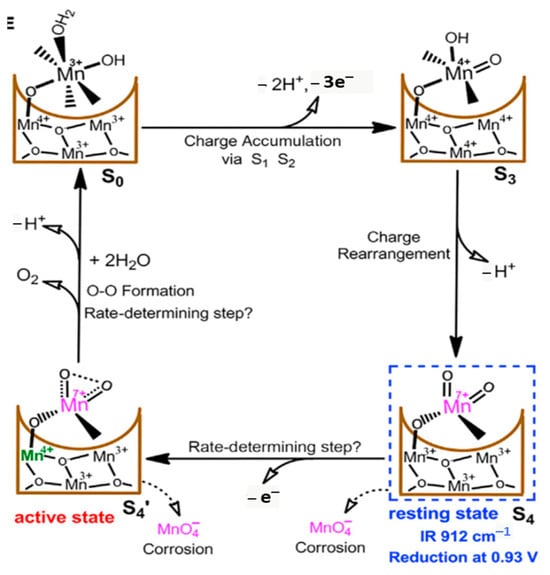
7.5. Mixed Metal Oxides
Two or three transition metals are mixed to form oxides of high electrocatalytic performance for water electrolyzers at a low cost. An NiFe oxide catalyst was employed as the anode catalyst with an NiMo oxide cathode catalyst with a high-performance perovskite-Si tandem solar cell, achieving a record 20% STH efficiency [105]. Nickel ferrite, NiFe2O4, cobalt ferrite, and CoFe2O4, are efficient and promising anode catalyst materials in the field of electrochemical water splitting. In Ni-Fe water oxidation electrocatalysts, Ni is likely not an active site for water oxidation, because Ni cannot achieve high-oxide states in aqueous environments at relevant potentials [106]. For the OER of NiFeOxHy, the addition of Co2+ cation increased the current density by 32.7% by the cation transport effect [107]. Using operando XAS, it was revealed that Ni oxidizes from the initial +2 oxidation state to the +3/+4 state [108]. For Ni-Fe oxyhydroxides, in situ monitoring of the Fe active site number and turn-frequency number provided important insights into the activity degradation/regeneration caused by Fe dissolution/adsorption as well as site-dependent activity and stability [109]. In the case of NiFe2O4, an Fe-site-assisted LOM pathway as the preferred OER mechanism was predicted. On the other hand, in the case of CoFe2O4, an Fe-site-assisted LOM pathway and a Co-site-assisted AEM pathway could both play a role [110]. Amorphous/crystalline NiFe2O4 induced by vanadium doping showed a superior electrocatalyst and long-term stability [111]. For amorphous Ni-Fe mixed metal oxides, analysis of the XAS revealed local structural transitions. A dual-site OER reaction mechanism was proposed, in which potential and rate-determining steps occur at Ni and Fe sites, respectively [112]. An Fe/Ca-based bimetallic oxide, CaFe2O4, exhibited outstanding OER activity in alkaline media. DFT calculations suggested an unconventional mechanism via the direct formation of O–O bonds between two oxygen intermediates, which are adsorbed on a multi-iron site on the catalyst surface [113]. On spinel NiCo2O4 abundant Co defects were preferentially produced by tuning the M–O bond length. Theoretical calculations and experiments proved that Al doping elongated the Co–O bond and promoted the ionization of Co under plasma treatment [114]. Spinel Co2MnO4 showed higher OER activity, most probably due to the ideal binding energies of the OER intermediates [115]. For modulated NiFeX and FeCoX (X = W, Mo, Nb, Ta, Re, and MoW) oxyhydroxide catalysts, in situ and ex situ soft and hard XAS were used to characterize the oxidation transition and facilitate the lower OER overpotential [116]. (Co–Fe–Pb)Ox in acidic solutions through a cobalt-selective self-healing mechanism was investigated. The kinetics of the process were investigated by soft XAS and it was revealed that low concentrations of Co2+ in the solution stabilize the catalytically active Co(Fe) sites [117].8. Layered Double Hydroxide (LDH)
LDH are emerging catalyst materials with inner layer water molecules and higher anion exchange capacity. They have been extensively used as electrocatalytic materials owing to their high specific surface area, environmental friendliness, lower cost, and non-toxicity [118]. A kind of LDH itself may become photocatalysts for water splitting. The electronic properties, such as band structure, bandgap energy (Eg), density of states (DOS), and band edge placement for MIIMIII-LDHs (MIII = Mg, Co, Ni and Zn; MIII = Al and Ga) were calculated by using the DFT + U method. The band structures of Mg- and Zn-based LDHs and Co- and Ni-based LDHs are responsive to ultraviolet (Eg > 3.1 eV) and visible light (Eg < 3.1 eV), respectively. The DOS calculations revealed that the photogenerated hole localizes on the surface hydroxyl group of LDHs, facilitating the oxidization of a water molecule without a long transportation route. The band edge placements of NiGa-, CoAl-, ZnAl-, and NiAl-LDHs have a driving force (0.965 eV, 0.836 eV, 0.667 eV, and 0.426 eV, respectively), toward oxygen evolution. In the experimental observations, only CoAl-LDH was an efficient oxygen evolution photocatalyst, agreeing well with the theoretical prediction [119].
References
- Wang, D.; Sheng, T.; Chen, J.; Wang, H.-F.; Hu, P. Identifying the key obstacle in photocatalytic oxygen evolution on rutile TiO2. Nat. Catal. 2018, 1, 291–299.
- Li, B.; Wu, S.; Gao, X. Theoretical calculation of a TiO2-based photocatalyst in the field of water splitting: A review. Nanotechnol. Rev. 2020, 9, 85.
- Malik, A.S.; Fredin, L.A. Unraveling the water oxidation mechanism on a stoichiometric and reduced rutile TiO2(100) surface using first-principles calculations. J. Phys. Chem. C 2023, 127, 3444–3451.
- Nosaka, Y.; Nosaka, A.Y. Generation and detection of reactive oxygen species in photocatalysis. Chem. Rev. 2017, 117, 11302–11336.
- Nosaka, Y.; Nosaka, A. Introduction to Photocatalysis: From Basic Science to Applications; Royal Society of Chemistry: Cambridge, UK, 2016; 272p.
- Kakuma, Y.; Nosaka, A.Y.; Nosaka, Y. Difference of TiO2 photocatalytic mechanism between rutile and anatase studied by the detection of active oxygen and surface species in water. Phys. Chem. Chem. Phys. 2015, 17, 18691–18698.
- Li, F.; Chen, J.-F.; Gong, X.-Q.; Hu, P.; Wang, D. Subtle structure matters: The vicinity of surface Ti5C cations alters the photooxidation behaviors of anatase and rutile TiO2 under aqueous environments. ACS Catal. 2022, 12, 8242–8251.
- Nosaka, Y. Water photo-oxidation over TiO2—History and reaction mechanism. Catalysts 2022, 12, 1557.
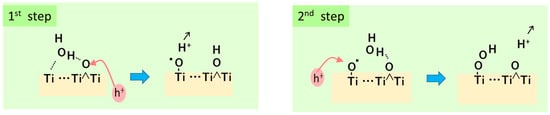
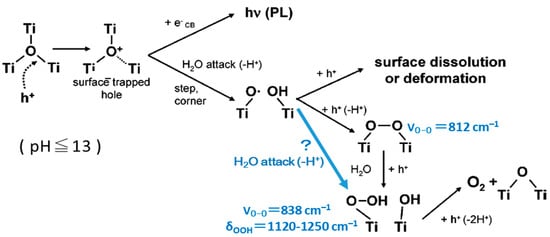
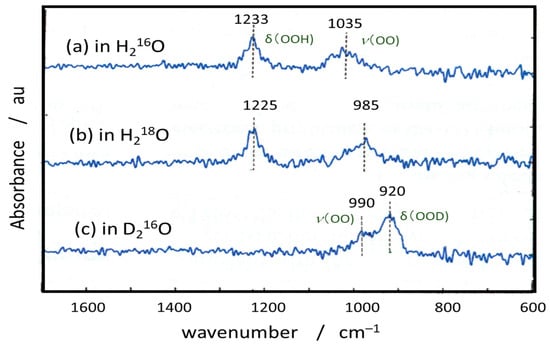
- Nakamura, R.; Nakato, Y. Primary intermediates of oxygen photoevolution reaction on TiO2 (rutile) particles, revealed by in situ FTIR absorption and photoluminescence measurements. J. Am. Chem. Soc. 2004, 126, 1290–1298.
- Nakamura, R.; Okamura, T.; Ohashi, N.; Imanishi, A.; Nakato, Y. Molecular mechanisms of photoinduced oxygen evolution, PL emission, and surface roughening at atomically smooth (110) and (100) n-TiO2 (rutile) surfaces in aqueous acidic solutions. J. Am. Chem. Soc. 2005, 127, 12975–12983.
- Nakamura, R.; Imanishi, A.; Murakoshi, K.; Nakato, Y. In situ FTIR studies of primary intermediates of photocatalytic reactions on nanocrystalline TiO2 films in contact with aqueous solutions. J. Am. Chem. Soc. 2003, 125, 7443–7450.
- Zhuang, Y.-B.; Cheng, J. Deciphering the anomalous acidic tendency of terminal water at rutile(110)–water interfaces. J. Phys. Chem. C 2023, 127, 10532–10540.
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278.
- Wang, Q.; Hisatomi, T.; Jia, Q.; Tokudome, H.; Zhong, M.; Wang, C.; Pan, Z.; Takata, T.; Nakabayashi, M.; Shibata, N.; et al. Scalable water splitting on particulate photocatalyst sheets with a solar-to-hydrogen energy conversion efficiency exceeding 1. Nat. Mater 2016, 15, 611–615.
- Rather, R.A.; Mehta, A.; Lu, Y.; Valant, M.; Fang, M.; Liu, W. Influence of exposed facets, morphology and hetero-interfaces of BiVO4 on photocatalytic water oxidation: A review. Int. J. Hydrogen Energy 2021, 46, 21866–21888.
- Ma, Y.; Pendlebury, S.R.; Reynal, A.; Formal, F.L.; Durrant, J.R. Dynamics of photogenerated holes in undoped BiVO4 photoanodes for solar water oxidation. Chem. Sci. 2014, 5, 2964–2973.
- Abdellaoui, I.; Islam, M.M.; Remeika, M.; Higuchi, Y.; Kawaguchi, T.; Harada, T.; Budich, C.; Maeda, T.; Wada, T.; Ikeda, S.; et al. Photocarrier recombination dynamics in BiVO4 for visible light-driven water oxidation. J. Phys. Chem. C 2020, 124, 3962–3972.
- Tran-Phu, T.; Fusco, Z.; Bernardo, I.D.; Lipton-Duffin, J.; Toe, C.Y.; Daiyan, R.; Gengenbach, T.; Lin, C.-H.; Bo, R.; Nguyen, H.T.; et al. Understanding the role of vanadium vacancies in BiVO4 for efficient photoelectrochemical water oxidation. Chem. Mater. 2021, 33, 3553–3565.
- Walsh, A.; Yan, Y.; Huda, M.N.; Al-Jassim, M.M.; Wei, S.-H. Band edge electronic structure of BiVO4: Elucidating the role of the Bi s and V d orbitals. Chem. Mater. 2009, 21, 547–551.
- Hu, J.; Zhao, X.; Chen, W.; Su, H.; Chen, Z. Theoretical insight into the mechanism of photoelectrochemical oxygen evolution reaction on BiVO4 anode with oxygen vacancy. J. Phys. Chem. C 2017, 121, 18702–18709.
- Liu, G.; Li, F.; Zhu, Y.; Lia, J.L.; Sun, L. Cobalt doped BiVO4 with rich oxygen vacancies for efficient photoelectrochemical water oxidation. RSC Adv. 2020, 10, 28523–28526.
- Liu, T.; Liu, R.; Li, Q.; Yang, J. Theoretical insight into the role of defects and facets in the selectivity of products in water oxidation over bismuth vanadate (BiVO4). ACS Sustain. Chem. Eng. 2020, 8, 1980–1988.
- Wang, W.; Strohbeen, P.J.; Lee, D.; Zhou, C.; Kawasaki, J.K.; Choi, K.-S.; Liu, M.; Galli, G. The role of surface oxygen vacancies in BiVO4. Chem. Mater. 2020, 32, 2899–2909.
- Steinitz-Eliyahu, R.; Hernangómez-Pérez, D.; Hegner, F.S.; Nikačević, P.; López, N.; Refaely-Abramson, S. Mixed excitonic nature in water-oxidized BiVO4 surfaces with defects. Phys. Rev. Mater. 2022, 6, 065402.
- Huang, M.; He, W.; Xu, Z.; Zhu, H. Enhanced catalytic mechanism of twin-structured BiVO4. J. Phys. Chem. Lett. 2021, 12, 10610–10615.
- Nikačević, P.; Hegner, F.S.; Galán-Mascarós, J.R.; López, N. Influence of oxygen vacancies and surface facets on water oxidation selectivity toward oxygen or hydrogen peroxide with BiVO4. ACS Catal. 2021, 11, 13416–13422.
- Park, H.S.; Leonard, K.C.; Bard, A.J. Surface interrogation scanning electrochemical microscopy (SISECM) of photoelectrochemistry at a W/Mo-BiVO4 semiconductor electrode. J. Phys. Chem. C 2013, 117, 12093–12102.
- Nakabayashi, Y.; Nishikawa, M.; Saito, N.; Terashima, C.; Fujishima, A. Significance of hydroxyl radical in photoinduced oxygen evolution in water on monoclinic bismuth vanadate. J. Phys. Chem. C 2017, 121, 25624–25631.
- Nakabayashi, Y.; Suzuki, N.; Terashima, C.; Fujishima, A. In situ infrared analysis for the process of water photo-oxidation on monoclinic bismuth vanadate. J. Phys. Chem. C 2021, 125, 18579–18587.
- Sun, Q.; Ren, K.; Qi, L. Boosting the performance of BiVO4 photoanodes by the simultaneous introduction of oxygen vacancies and cocatalyst via photoelectrodeposition. ACS Appl. Mater. Interfaces 2022, 14, 37833–37842.
- Pan, L.; Wu, J.; Xu, X.; Lv, F.; Chen, Y.; Guo, L. Photoelectrochemical performance of bismuth vanadate photoanode for water splitting under concentrated light irradiation. Int. J. Hydrogen Energy 2023, 48, 13479–13488.
- Majumder, S.; Su, X.; Kim, K.H. Effective strategy of incorporating Co3O4 as a co-catalyst onto an innovative BiVO4/Fe2TiO5 core-shell heterojunction for effective photoelectrochemical water-splitting application. Surf. Interfaces. 2023, 39, 102936.
- Avcıoǧlu, C.; Avcıoǧlu, S.; Bekheet, M.F.; Gurlo, A. Photocatalytic overall water splitting by SrTiO3: Progress report and design strategies. ACS Appl. Energy Mater. 2023, 6, 1134–1154.
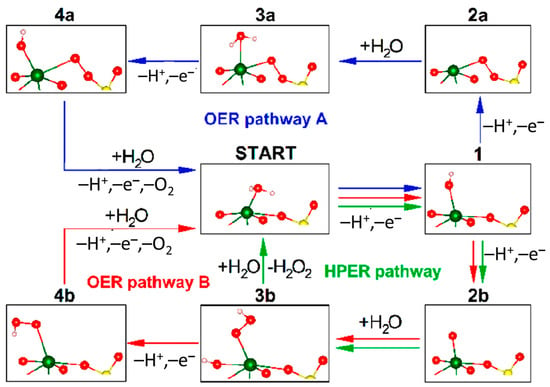
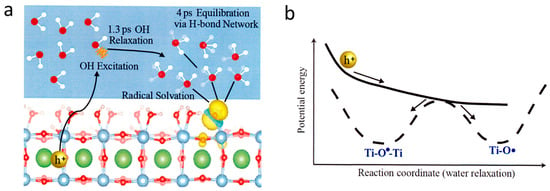
- Chen, X.; Choing, S.N.; Aschaffenburg, D.J.; Pemmaraju, C.D.; Prendergast, D.; Cuk, T. The formation time of Ti–O• and Ti–O•–Ti radicals at the n-SrTiO3/aqueous interface during photocatalytic water oxidation. J. Am. Chem. Soc. 2017, 139, 1830–1841.
- Chen, X.; Aschaffenburg, D.J.; Cuk, T. Selecting between two transition states by which water oxidation intermediates decay on an oxide surface. Nat. Catal. 2019, 2, 820–827.
- Vinogradov, I.; Singh, S.; Lyle, H.; Paolino, M.; Mandal, A.; Rossmeisl, J.; Cuk, T. Free energy difference to create the M-OH• intermediate of the oxygen evolution reaction by time-resolved optical spectroscopy. Nat. Mater. 2022, 21, 88–94.
- Lyle, H.; Singh, S.; Magnano, E.; Nappini, S.; Bondino, F.; Yazdi, S.; Cuk, T. Assessing and quantifying thermodynamically concomitant degradation during oxygen evolution from water on SrTiO3. ACS Catal. 2023, 13, 8206–8218.
- Chen, T.; Ding, Q.; Wang, X.; Feng, Z.; Li, C. Mechanistic studies on photocatalytic overall water splitting over Ga2O3-based photocatalysts by operando MS-FTIR spectroscopy. J. Phys. Chem. Lett. 2021, 12, 6029–6033.
- Zhou, X.; Hensen, E.J.M.; van Santen, R.A.; Li, C. DFT simulations of water adsorption and activation on low-index α-Ga2O3 surfaces. Chem. Eur. J. 2014, 20, 6915–6926.
- Wang, Q.; Nakabayashi, M.; Hisatomi, T.; Sun, S.; Akiyama, S.; Wang, Z.; Pan, Z.; Xiao, X.; Watanabe, T.; Yamada, T.; et al. Oxysulfide photocatalyst for visible-light-driven overall water splitting. Nat. Mater. 2019, 18, 827–832.
- Yu, H.; Ge, J. Recent advances in Ru-based electrocatalysts toward acid electrochemical water oxidation. Curr. Opinion Electrochem. 2023, 39, 101296.
- Raman, A.S.; Vojvodic, A. Providing atomistic insights into the dissolution of rutile oxides in electrocatalytic water splitting. J. Phys. Chem. C 2022, 126, 922–932.
- Liu, S.; Chang, Y.; He, N.; Zhu, S.; Wang, L.; Liu, X. Competition between lattice oxygen and adsorbate evolving mechanisms in rutile Ru-based oxide for the oxygen evolution reaction. ACS Appl. Mater. Interf. 2023, 15, 20563–20570.
- Pavlovic, Z.; Ranjan, C.; Gao, Q.; van Gastel, M.; Schlögl, R. Probing the structure of a water-oxidizing anodic iridium oxide catalyst using Raman spectroscopy. ACS Catal. 2016, 6, 8098–8105.
- Sivasankar, N.; Weare, W.W.; Frei, H. Direct observation of a hydroperoxide surface intermediate upon visible light-driven water oxidation at an Ir oxide nanocluster catalyst by rapid-scan FT-IR spectroscopy. J. Am. Chem. Soc. 2011, 133, 12976–12979.
- Ooka, H.; Yamaguchi, A.; Takashima, T.; Hashimoto, K.; Nakamura, R. Efficiency of oxygen evolution on iridium oxide determined from the pH dependence of charge accumulation. J. Phys. Chem. C 2017, 121, 17873–17881.
- Bozal-Ginesta, C.; Rao, R.R.; Mesa, C.A.; Liu, X.; Hillman, S.A.J.; Stephens, I.E.L.; Durrant, J.R. Redox-state kinetics in water-oxidation IrOX electrocatalysts measured by operando spectroelectrochemistry. ACS Catal. 2021, 11, 15013–15025.
- Czioska, S.; Boubnov, A.; Escalera-López, D.; Geppert, J.; Zagalskaya, A.; Röse, P.; Saraçi, E.; Alexandrov, V.; Krewer, U.; Cherevko, S.; et al. Increased Ir−Ir interaction in iridium oxide during the oxygen evolution reaction at high potentials probed by operando spectroscopy. ACS Catal. 2021, 11, 10043–10057.
- Ping, Y.; Nielsen, R.J.; Goddard III, W.A. The reaction mechanism with free energy barriers at constant potentials for the oxygen evolution reaction at the IrO2(110) surface. J. Am. Chem. Soc. 2017, 139, 149–155.
- Binninger, T.; Doublet, M.-L. The Ir–OOOO–Ir transition state and the mechanism of the oxygen evolution reaction on IrO2(110). Energy Environ. Sci. 2022, 15, 2519–2528.
- Liao, F.; Yin, K.; Ji, Y.; Zhu, W.; Fan, Z.; Li, Y.; Zhong, J.; Shao, M.; Kang, Z.; Shao, Q. Iridium oxide nanoribbons with metastable monoclinic phase for highly efficient electrocatalytic oxygen evolution. Nat. Commun. 2023, 14, 1248.
- Xie, Y.; Chang, C.; Luo, F.; Yang, Z. Modulation in the d band of Ir by core–shell construction for robust water splitting electrocatalysts in acid. ACS Appl. Mater. Interfaces. 2023, 15, 20081–20088.
- Reksten, A.H.; Thuv, H.; Seland, F.; Sunde, S. The oxygen evolution reaction mechanism at IrXRu1−XO2 powders produced by hydrolysis synthesis. J. Electroanal. Chem. 2018, 819, 547–561.
- Rao, R.R.; Kolb, M.J.; Halck, N.B.; Pedersen, A.F.; Mehta, A.; You, H.; Stoerzinger, K.A.; Feng, Z.; Hansen, H.A.; Zhou, H.; et al. Towards identifying the active sites on RuO2(110) in catalyzing oxygen evolution. Energy Environ. Sci. 2017, 10, 2626–2637.
- Liang, Q.; Bieberle-Hütter, A.; Brocks, G. Anti-ferromagnetic RuO2: A stable and robust OER catalyst over a large range of surface terminations. J. Phys. Chem. C 2022, 126, 1337–1345.
- Stoerzinger, K.A.; Diaz-Morales, O.; Kolb, M.; Rao, R.R.; Frydendal, R.; Qiao, L.; Wang, X.R.; Halck, N.B.; Rossmeisl, J.; Hansen, H.A.; et al. Orientation-dependent oxygen evolution on RuO2 without lattice exchange. ACS Energy Lett. 2017, 2, 876–881.
- Wang, Y.; Yang, R.; Ding, Y.; Zhang, B.; Li, H.; Bai, B.; Li, M.; Cui, Y.; Xiao, J.; Wu, Z.-S. Unraveling oxygen vacancy site mechanism of Rh-doped RuO2 catalyst for long-lasting acidic water oxidation. Nat. Commun. 2023, 14, 1412.
- Shang, C.; Xiao, X.; Xu, Q. Coordination chemistry in modulating electronic structures of perovskite-type oxide nanocrystals for oxygen evolution catalysis. Coord. Chem. Rev. 2023, 485, 215109.
- Hong, W.T.; Stoerzinger, K.A.; Lee, Y.-L.; Giordano, L.; Grimaud, A.; Johnson, A.M.; Hwang, J.; Crumlin, E.J.; Yange, W.; Shao-Horn, Y. Charge-transfer-energy-dependent oxygen evolution reaction mechanisms for perovskite oxides. Energy Environ. Sci. 2017, 10, 2190–2200.
- Shi, Z.; Wang, X.; Ge, J.; Liu, C.; Xing, W. Fundamental understanding of the acidic oxygen evolution reaction: Mechanism study and state-of-the-art catalysts. Nanoscale 2020, 12, 13249–13275.
- Lu, M.; Zheng, Y.; Hu, Y.; Huang, B.; Ji, D.; Sun, M.; Li, J.; Peng, Y.; Si, R.; Xi, P.; et al. Artificially steering electrocatalytic oxygen evolution reaction mechanism by regulating oxygen defect contents in perovskites. Sci. Adv. 2022, 8, eabq3563.
- Peng, M.; Huang, J.; Zhu, Y.; Zhou, H.; Hu, Z.; Liao, Y.-K.; Lai, Y.-H.; Chen, C.-T.; Chu, Y.-H.; Zhang, K.H.L.; et al. Structural anisotropy determining the oxygen evolution mechanism of strongly correlated perovskite nickelate electrocatalyst. ACS Sustain. Chem. Eng. 2021, 9, 4262–4270.
- Sun, Y.; Wu, C.-R.; Ding, T.-Y.; Gu, J.; Yan, J.-W.; Cheng, J.; Zhang, K.H.L. Direct observation of the dynamic reconstructed active phase of perovskite LaNiO3 for the oxygen evolution reaction. Chem. Sci. 2023, 14, 5906–5911.
- Lee, S.; Kishore, M.R.A.; Kim, D.; Kang, H.; Chun, J.; Oh, L.S.; Park, J.H.; Kim, H.J.; Yoo, J.S.; Lim, E. Direct O–O coupling promoted the oxygen evolution reaction by dual active sites from Ag/LaNiO3 interfaces. ACS Appl. Energy Mater. 2022, 5, 14658–14668.
- Li, N.; Cai, L.; Gao, G.; Lin, Y.; Wang, C.; Liu, H.; Liu, Y.; Duan, H.; Ji, Q.; Hu, W.; et al. Operando direct observation of stable water-oxidation intermediates on Ca2−XIrO4 nanocrystals for efficient acidic oxygen evolution. Nano Lett. 2022, 22, 6988–6996.
- Chen, H.; Shi, L.; Sun, K.; Zhang, K.; Liu, Q.; Ge, J.; Liang, X.; Tian, B.; Huang, Y.; Shi, Z.; et al. Protonated iridate nanosheets with a highly active and stable layered perovskite framework for acidic oxygen evolution. ACS Catal. 2022, 12, 8658–8666.
- Yu, M.; Budiyanto, E.; Tgysgz, H. Principles of water electrolysis and recent progress in cobalt-, nickel-, and iron-based oxides for the oxygen evolution reaction. Angew. Chem. Int. Ed. 2022, 61, e202103824.
- Han, J.; Guan, J. Multicomponent transition metal oxides and (oxy)hydroxides for oxygen evolution. Nano Res. 2023, 16, 1913–1966.
- Wang, H.; Zhang, K.H.L.; Hofmann, J.P.; O’Shea, V.A.; Oropeza, F.E. The electronic structure of transition metal oxides for oxygen evolution reaction. J. Mater. Chem. A 2021, 9, 19465–19488.
- Feng, Y.; Yang, H.; Wang, X.; Hu, C.; Jing, H.; Cheng, J. Role of transition metals in catalyst designs for oxygen evolution reaction: A comprehensive review. Int. J. Hydrogen Energy 2022, 47, 17946–17970.
- Guo, T.; Li, L.; Wang, Z. Recent development and future perspectives of amorphous transition metal-based electrocatalysts for oxygen evolution reaction. Adv. Energy Mater. 2022, 12, 2200827.
- Huang, C.-J.; Xu, H.-M.; Shuai, T.-Y.; Zhan, Q.-N.; Zhang, Z.-J.; Li, G.-R. A review of modulation strategies for improving catalytic performance of transition metal phosphides for oxygen evolution reaction. App. Catal. B Environ. 2023, 325, 122313.
- Zhang, W.; Cao, R. Switching the O–O bond formation mechanism by controlling water activity. Chem 2021, 7, 1981–1992.
- Takata, T.; Jiang, J.; Sakata, Y.; Nakabayashi, M.; Shibata, N.; Nandal, V.; Seki, K.; Hisatomi, T.; Domen, K. Photocatalytic water splitting with a quantum efficiency of almost unity. Nature 2020, 581, 411–414.
- Wang, C.; Deng, R.; Guo, M.; Zhang, Q. Recent progress of advanced Co3O4-based materials for electrocatalytic oxygen evolution reaction in acid: From rational screening to efficient design. Int. J. Hydrogen Energy, 2023; in press.
- Favaro, M.; Yang, J.; Nappini, S.; Magnano, E.; Toma, F.M.; Crumlin, E.J.; Yano, J.; Sharp, I.D. Understanding the oxygen evolution reaction mechanism on CoOX using operando ambient-pressure X-ray photoelectron spectroscopy. J. Am. Chem. Soc. 2017, 139, 8960–8970.
- Moysiadou, A.; Lee, S.; Hsu, C.-S.; Chen, H.M.; Hu, X. Mechanism of oxygen evolution catalyzed by cobalt oxyhydroxide: Cobalt superoxide species as a key intermediate and dioxygen release as a rate-determining step. J. Am. Chem. Soc. 2020, 142, 11901–11914.
- Lang, C.; Li, J.; Yang, K.R.; Wang, Y.; He, D.; Thorne, J.E.; Croslow, S.; Dong, Q.; Zhao, Y.; Prostko, G.; et al. Observation of a potential-dependent switch of water-oxidation mechanism on Co-oxide-based catalysts. Chem 2021, 7, 2101–2117.
- Wang, S.; Jiang, Q.; Ju, S.; Hsu, C.-S.; Chen, H.M.; Zhang, D.; Song, F. Identifying the geometric catalytic active sites of crystalline cobalt oxyhydroxides for oxygen evolution reaction. Nat. Commun. 2022, 13, 6650.
- Kang, W.; Wei, R.; Yin, H.; Li, D.; Chen, Z.; Huang, Q.; Zhang, P.; Jing, H.; Wang, X.; Li, C. Unraveling sequential oxidation kinetics and determining roles of multi-cobalt active sites on Co3O4 catalyst for water oxidation. J. Am. Chem. Soc. 2023, 145, 3470–3477.
- Lin, Y.; Yu, L.; Tang, L.; Song, F.; Schlögl, R.; Heumann, S. In situ identification and time-resolved observation of the interfacial state and reactive intermediates on a cobalt oxide nanocatalyst for the oxygen evolution reaction. ACS Catal. 2022, 12, 5345–5355.
- Zhou, D.; Li, F.; Zhao, Y.; Wang, L.; Zou, H.; Shan, Y.; Fu, J.; Ding, Y.; Duan, L.; Liu, M.; et al. Mechanistic regulation by oxygen vacancies in structural evolution promoting electrocatalytic water oxidation. ACS Catal. 2023, 13, 4398–4408.
- Ren, X.; Ji, Y.; Zhai, Y.; Yuan, N.; Ding, J.; Li, Y.; Yan, J.; Liu, S.F. Self-assembled CoOOH on TiO2 for enhanced photoelectrochemical water oxidation. J. Energy Chem. 2021, 60, 512–521.
- Yao, N.; Wang, G.; Jia, H.; Yin, J.; Cong, H.; Chen, S.; Luo, W. Intermolecular energy gap-induced formation of high-valent cobalt species in CoOOH surface layer on cobalt sulfides for efficient water oxidation. Angew. Chem. Int. Ed. 2022, 61, e202117178.
- Li, L.-F.; Li, Y.-F.; Liu, Z.-P. Oxygen evolution activity on NiOOH catalysts: Four-coordinated Ni cation as the active site and the hydroperoxide mechanism. ACS Catal. 2020, 10, 2581–2590.
- Wang, X.; Xi, S.; Huang, P.; Du, Y.; Zhong, H.; Wang, Q.; Borgna, A.; Zhang, Y.-W.; Wang, Z.; Wang, H.; et al. Pivotal role of reversible NiO6 geometric conversion in oxygen evolution. Nature 2022, 611, 702–708.
- Wang, H.; Lu, S. Light inducing the geometric conversion of NiO6 to trigger a faster oxygen evolution reaction pathway: The coupled oxygen evolution mechanism. Energy Environ. Mater. 2023, 6, e12558.
- Jiang, J.; Sun, F.; Zhou, S.; Hu, W.; Zhang, H.; Dong, J.; Jiang, Z.; Zhao, J.; Li, J.; Yan, W.; et al. Atomic-level insight into super-efficient electrocatalytic oxygen evolution on iron and vanadium co-doped nickel (oxy)hydroxide. Nat. Commun. 2018, 9, 2885.
- Rao, R.R.; Corby, S.; Bucci, A.; García-Tecedor, M.; Mesa, C.A.; Rossmeisl, J.; Giménez, S.; Lloret-Fillol, J.; Stephens, I.E.L.; Durrant, J.R. Spectroelectrochemical analysis of the water oxidation mechanism on doped nickel oxides. J. Am. Chem. Soc. 2022, 144, 7622–7633.
- Li, T.; Zhang, L.; Wang, J.; Zhang, X.; Zhang, L.; Wang, M.; Yan, C.; Tao Qian, T. Facilitating reconstruction of the hetero interface electronic structure by the enriched oxygen vacancy for the oxygen evolution reaction. Inorg. Chem. 2023, 62, 10504–10512.
- Liu, J.; Xiao, J.; Wang, Z.; Yuan, H.; Lu, Z.; Luo, B.; Tian, E.; Waterhouse, G.I.N. Structural and electronic engineering of Ir-doped Ni-(oxy)hydroxide nanosheets for enhanced oxygen evolution activity. ACS Catal. 2021, 11, 5386–5395.
- Tian, B.; Shin, H.; Liu, S.; Fei, M.; Mu, Z.; Liu, C.; Pan, Y.; Sun, Y.; Goddard III, W.A.; Ding, M. Double-exchange-induced in situ conductivity in nickel-based oxyhydroxides: An effective descriptor for electrocatalytic oxygen evolution. Angew. Chem. Int. Ed. 2021, 60, 16448–16456.
- Xiong, Y.; He, P. A review on electrocatalysis for alkaline oxygen evolution reaction (OER) by Fe-based catalysts. J. Mater. Sci. 2023, 58, 2041–2067.
- Zhang, Y.; Zhang, H.; Liu, A.; Chen, C.; Song, W.; Zhao, J. Rate-limiting O–O bond formation pathways for water oxidation on hematite photoanode. J. Am. Chem. Soc. 2018, 140, 3264–3269.
- Righi, G.; Plescher, J.; Schmidt, F.-P.; Campen, R.K.; Fabris, S.; Knop-Gericke, A.; Schlögl, R.; Jones, T.E.; Teschner, D.; Piccinin, S. On the origin of multihole oxygen evolution in haematite photoanodes. Nat. Catal. 2022, 5, 888–899.
- Adak, M.K.; Mallick, L.; Samanta, K.; Chakraborty, B. Slow O–H dissociation in the first-order oxygen evolution reaction kinetics on polycrystalline γ-FeO(OH). J. Phys. Chem. C 2023, 127, 154–168.
- Gao, L.; Tang, C.; Liu, J.; He, L.; Wang, H.; Ke, Z.; Li, W.; Jiang, C.; He, D.; Cheng, L.; et al. Oxygen vacancy-induced electron density tuning of Fe3O4 for enhanced oxygen evolution catalysis. Energy Environ. Mater. 2021, 4, 392–398.
- Ouyang, Q.; Cheng, S.; Yang, C.; Lei, Z. Ni, Co, and Yb cation Co-doping and defect engineering of FeOOH nanorods as an electrocatalyst for the oxygen evolution reaction. Inorg. Chem. 2023, 62, 1719–1727.
- Li, C.; Chen, M.; Xie, Y.; Wang, H.; Jia, L. Boosting photoelectrochemical water splitting of bismuth vanadate photoanode via novel co-catalysts of amorphous manganese oxide with variable valence states. J. Colloid. Interf. Sci. 2023, 636, 103–112.
- Zahran, Z.N.; Mohamed, E.A.; Naruta, Y. Kinetic and mechanism of heterogeneous water oxidation by α-Mn2O3 sintered on FTO electrode. ACS Catal. 2016, 6, 4470–4476.
- Mousazade, Y.; Mohammadi, M.R.; Chernev, P.; Bikas, R.; Song, Z.; Lis, T.; Dau, H.; Najafpour, M.M. Water oxidation by a manganese–potassium cluster: Mn oxide as a kinetically dominant “true” catalyst for water oxidation. Catal. Sci. Technol. 2018, 8, 4390–4398.
- Seo, H.; Park, S.; Cho, K.H.; Choi, S.; Ko, C.; Randriamahazaka, H.; Nam, K.T. Complex impedance analysis on charge accumulation step of Mn3O4 nanoparticles during water oxidation. ACS Omega 2021, 6, 18404–18413.
- Yoon, S.; Seo, H.; Jin, K.; Kim, H.G.; Lee, S.-Y.; Jo, J.; Cho, K.H.; Ryu, J.; Yoon, A.; Kim, Y.-W.; et al. Atomic reconstruction and oxygen evolution reaction of Mn3O4 nanoparticles. J. Phys. Chem. Lett. 2022, 13, 8336–8343.
- Zhang, B.; Daniel, Q.; Fan, L.; Liu, T.; Meng, Q.; Sun, L. Identifying MnVII-oxo species during electrochemical water oxidation by manganese oxide. iScience 2018, 4, 144–152.
- Wang, Y.; Sharma, A.; Duong, T.; Arandiyan, H.; Zhao, T.; Zhang, D.; Su, Z.; Garbrecht, M.; Beck, F.J.; Karuturi, S.; et al. Direct solar hydrogen generation at 20% efficiency using low-cost materials. Adv. Energy Mat. 2021, 11, 2101053.
- Zhang, J.; Winkler, J.R.; Gray, H.B.; Hunter, B.M. Mechanism of nickel−iron water oxidation electrocatalysts. Energy Fuels 2021, 35, 19164–19169.
- Yang, Y.; Du, X.; Wang, S.; Zhao, K.; Wang, L.; Qi, Z.; Yang, W.; Hao, J.; Shi, W. Cation transport effect on nickel iron oxyhydroxide electrodes in the oxygen evolution reaction. Ind. Eng. Chem. Res. 2022, 61, 16702–16710.
- Acharya, P.; Manso, R.H.; Hoffman, A.S.; Bakovic, S.I.P.; Kékedy-Nagy, L.; Bare, S.R.; Chen, J.; Greenlee, L.F. Fe coordination environment, Fe-incorporated Ni(OH) phase, and metallic core are key structural components to active and stable nanoparticle catalysts for the oxygen evolution reaction. ACS Catal. 2022, 12, 1992–2008.
- Qian, H.; Wei, J.; Yu, C.; Tang, F.; Jiang, W.; Xia, D.; Gan, L. In situ quantification of the active sites, turnover frequency, and stability of Ni–Fe (oxy)hydroxides for the oxygen evolution reaction. ACS Catal. 2022, 12, 14280–14289.
- Avcı, O.N.; Sementa, L.; Fortunelli, A. Mechanisms of the oxygen evolution reaction on NiFe2O4 and CoFe2O4 inverse-spinel oxides. ACS Catal. 2022, 12, 9058–9073.
- Deng, H.; Jiang, H.; Wang, K.; Wang, Z.; Wang, B.; Zhou, Z.; Li, J. Coupling the vanadium-induced amorphous/crystalline NiFe2O4 with phosphide heterojunction toward active oxygen evolution reaction catalysts. Nanotechnol. Rev. 2022, 11, 3165–3173.
- Schoen, M.A.W.; Calderon, O.; Randell, N.M.; Jimenez-Villegas, S.; Daly, K.M.; Chernikov, R.; Trudel, S. Local structural changes in polyamorphous (Ni,Fe)O electrocatalysts suggest a dual-site oxygen evolution reaction mechanism. J. Mater. Chem. A 2021, 9, 13252–13262.
- Sugawara, Y.; Kamata, K.; Ishikawa, A.; Tateyama, Y.; Yamaguchi, T. Efficient oxygen evolution electrocatalysis on CaFe2O4 and its reaction mechanism. ACS Appl. Energy Mater. 2021, 4, 3057–3066.
- Zheng, J.; Peng, X.; Xu, Z.; Gong, J.; Wang, Z. Cationic defect engineering in spinel NiCo2O4 for enhanced electrocatalytic oxygen evolution. ACS Catal. 2022, 12, 10245–10254.
- Li, A.; Kong, S.; Guo, C.; Ooka, H.; Adachi, K.; Hashizume, D.; Jiang, Q.; Han, H.; Xiao, J.; Nakamura, R. Enhancing the stability of cobalt spinel oxide towards sustainable oxygen evolution in acid. Nat. Catal. 2022, 5, 109–118.
- Zhang, B.; Wang, L.; Cao, Z.; Kozlov, S.M.; de Arquer, F.P.G.; Dinh, C.T.; Li, J.; Wang, Z.; Zheng, X.; Zhang, L.; et al. High-valence metals improve oxygen evolution reaction performance by modulating 3d metal oxidation cycle energetics. Nat. Catal. 2020, 3, 985–992.
- Simondson, D.; Chatti, M.; Bonke, S.A.; Tesch, M.F.; Golnak, R.; Xiao, J.; Hoogeveen, D.A.; Cherepanov, P.V.; Gardiner, J.L.; Tricoli, A.; et al. Stable acidic water oxidation with a cobalt–iron–lead oxide catalyst operating via a cobalt-selective self-healing mechanism. Angew. Chem. Int. Ed. 2021, 60, 15821–15826.
- Karim, A.V.; Hassani, A.; Eghbali, P.; Nidheesh, P.V. Nanostructured modified layered double hydroxides (LDHs)-based catalysts: A review on synthesis, characterization, and applications in water remediation by advanced oxidation processes. Curr. Opinion Solid State and Mater. Sci. 2022, 26, 100965.
- Xu, S.-M.; Pan, T.; Dou, Y.-B.; Yan, H.; Zhang, S.-T.; Ning, F.-Y.; Shi, W.-Y.; Wei, M. Theoretical and experimental study on MIIMIII -layered double hydroxides as efficient photocatalysts toward oxygen evolution from water. J. Phys. Chem. C 2015, 119, 18823–18834.
- Zhang, H.; Wu, L.; Feng, R.; Wang, S.; Hsu, C.-S.; Ashfaq, Y.N.; Zhang, A.C.; Wu, H.; Chen, H.-M.; Zhang, W.; et al. Oxygen vacancies unfold the catalytic potential of NiFe-layered double hydroxides by promoting their electronic transport for oxygen evolution reaction. ACS Catal. 2023, 13, 6000–6012.
- Kang, J.; Qiu, X.; Hu, Q.; Zhong, J.; Gao, X.; Huang, R.; Wan, C.; Liu, L.-M.; Duan, X.; Guo, L. Valence oscillation and dynamic active sites in monolayer NiCo hydroxides for water oxidation. Nat. Catal. 2021, 4, 1050–1058.
- Lee, S.; Banjac, K.; Lingenfelder, M.; Hu, X. Oxygen isotope labeling experiments reveal different reaction sites for the oxygen evolution reaction on nickel and nickel iron oxides. Angew. Chem. Int. Ed. 2019, 58, 10295–10299.
- Wang, Z.; Goddard III, W.A.; Xiao, H. Potential-dependent transition of reaction mechanisms for oxygen evolution on layered double hydroxides. Nat. Commun. 2023, 14, 4228.
- Xu, J.; Li, Z.; Chen, D.; Yang, S.; Zheng, K.; Ruan, J.; Wu, Y.; Zhang, H.; Chen, J.; Xie, F.; et al. Active electrocatalyst in the oxygen evolution reaction and flexible zinc–air batteries. ACS Appl. Mater. Interfaces 2021, 13, 48774–48783.
- Sun, Z.; Lin, L.; He, J.; Ding, D.; Wang, T.; Li, J.; Li, M.; Liu, Y.; Li, Y.; Yuan, M.; et al. Regulating the spin state of FeIII enhances the magnetic effect of the molecular catalysis mechanism. J. Am. Chem. Soc. 2022, 144, 8204–8213.
- Qiao, C.; Usman, Z.; Wei, J.; Gan, L.; Hou, J.; Hao, Y.; Zhu, Y.; Zhang, J.; Cao, C. Efficient O–O coupling at catalytic interface to assist kinetics optimization on concerted and sequential proton–electron transfer for water oxidation. ACS Nano 2023, 17, 12278–12289.
- He, X.; Han, X.; Zhou, X.; Chen, J.; Wang, J.; Chen, Y.; Yu, L.; Zhang, N.; Li, J.; Wang, S.; et al. Electronic modulation with Pt-incorporated NiFe layered doublehydroxide for ultrastable overall water splitting at 1000 mA cm−2. App. Catal. B Environ. 2023, 331, 122683.
- Gao, Z.-W.; Liu, J.-Y.; Chen, X.-M.; Zheng, X.-L.; Mao, J.; Liu, H.; Ma, T.; Li, L.; Wang, W.-C.; Du, X.-W. Engineering NiO/NiFe LDH intersection to bypass scaling relationship for oxygen evolution reaction via dynamic tridimensional adsorption of intermediates. Adv. Mater. 2019, 31, 1804769.
- Luo, Y.; Wu, Y.; Wu, D.; Huang, C.; Xiao, D.; Chen, H.; Zheng, S.; Chu, P.K. NiFe-layered double hydroxide synchronously activated by heterojunctions and vacancies for the oxygen evolution reaction. ACS Appl. Mater. Interfaces 2020, 12, 42850–42858.
- Zhai, P.; Wang, C.; Zhao, Y.; Zhang, Y.; Gao, J.; Sun, L.; Hou, J. Regulating electronic states of nitride/hydroxide to accelerate kinetics for oxygen evolution at large current density. Nat. Commun. 2023, 14, 1873.
- Liang, X.; Li, Y.; Fan, H.; Deng, S.; Zhao, X.; Chen, M.; Pan, G.; Xiong, Q.; Xia, X. Bifunctional NiFe layered double hydroxide@Ni3S2 hetero structure as efficient electrocatalyst for overall water splitting. Nanotechnology 2019, 30, 484001.
- Wu, Z.; Liu, X.; Li, H.; Sun, Z.; Cao, M.; Li, Z.; Fang, C.; Zhou, J.; Cao, C.; Dong, J.; et al. A semiconductor-electrocatalyst nano interface constructed for successive photoelectrochemical water oxidation. Nat. Commun. 2023, 14, 2574.
- Guru, S.; Rao, G.R. Review—Strategic design of layered double hydroxides and graphitic carbon nitride heterostructures for photoelectrocatalytic water splitting applications. J. Electrochem. Soc. 2022, 169, 046515.