Amyotrophic Lateral Sclerosis (ALS) is a progressive motor neurodegenerative disease. Cell damage in ALS is the result of many different, largely unknown, pathogenetic mechanisms. Superoxide Dismutase 1 (SOD1) physiologically mediates intracellular peroxide generation. About 10% of ALS subjects show a familial disease associated with different gain-of-function SOD1 mutations. The occurrence of sporadic ALS, not clearly associated with SOD1 defects, has been also described. SOD1-dependent pathways have been involved in neuron functional network as well as in immune-response regulation.
- Amyotrophic Lateral Sclerosis (ALS)
- Superoxide Dismutase 1 (SOD1)
- intracellular signaling
1. Pathogenic Mechanisms of Mutant SOD1 in ALS
2. Microglia Activation, NADPH Oxidase and SOD1 in ALS
Activated microglia represents a key element in Alzheimer’s disease as well as in the pathogenesis of ALS and other neurodegenerative conditions [62,63,64][22][23][24]. Microglia activation can be observed in transgenic mice expressing human SOD1 mutants, before neuron loss [65][25]; thus, dysregulated microglia functions, together with astrocyte activation, carry out an important role in ALS pathogenesis/progression [62,63,64,66][22][23][24][26]. ROS generated from NADPH oxidases play a role in signaling events leading to microglia activation [67][27]. Seven structural homologues of the phagocyte NOX enzyme (NOX2) have been identified, such as NOX1-5, DUOX1 and DUOX2 [68][28]. On the other hand, activated microglia produces ROS, primarily by NADPH oxidase 2 (NOX2), that result in enhanced microglia activation. It has also been shown that redox distress, caused by NOX1 and NOX2, significantly influences the progression of motor neuron disease, in mutant SOD1G93A ALS mice [69][29]. Generation of ROS represents a general phenomenon in human cells. However, the excess of ROS production or an imbalance between ROS production and antioxidant defense are thought to represent important factors of disease progression in ALS [70,71][30][31]. Recent studies have shown that SOD1G93A ALS transgenic mice generate high levels of gp91PHOX (NOX2) and superoxide in spinal cord microglia [72][32]. Moreover, it has been proposed that SOD1 interacts directly with Rac1, a cytosolic regulator of NOX2 [73][33], resulting in overproduction of ROS. In addition, ROS generated by Rac-dependent NADPH oxidases have been observed to be involved in cell signaling as well as in microbial killing. Harraz [74][34] hypothesized that Rac-GTP-mediated activation of the NADPH oxidase complex might lead to production of O2•− and H2O2, which were able to mediate Nox complex autoregulation by reducing Rac-GTP levels. Interestingly, Marden [69][29] showed that female ALS mice, lacking a copy of the X-chromosomal Nox1 or Nox2 genes, exhibited significantly increased survival rates, thus suggesting that a 50% reduction in NOX1/NOX2 expression levels might be associated with a substantial improvement of ALS outcome in mice. Moreover, the observation that multiple Nox genes might contribute to ALS progression clearly expands the potential therapeutic targets for this disease.3. SOD1, Immunity and Neuroinflammation Processes in ALS
Chronic inflammation has been considered an important element in the pathogenesis/progression of different diseases, as well as in neurodegenerative processes. [75][35]. In this context, multiple immune dysfunctions, represented by extensive, dysregulated inflammatory processes, auto immunity phenomena, and deranged immune responses have been described in ALS. In addition, mutations in several genes, directly involved in immune response, have recently been reported in ALS patients [76,77,78,79][36][37][38][39]. Neuroinflammation, in particular, behaves as a key modulator of ALS progression potentially representing a prospective therapeutic target for this disease. In ALS, inflammatory responses are not restricted to the proximity of motoneurons but have been detected in muscles, peripheral axons, skin, liver and blood [80,81,82,83,84,85][40][41][42][43][44][45]. Multiple immune cell subsets have been described to participate in ALS pathogenetic mechanisms. Indeed, T cells, monocytes and other immune effectors have been observed to directly or indirectly access the CNS through the choroid plexus [86][46], thus mediating neuron damaging as well as neuroprotective processes. E. Coque et al. [87][47] showed that ablation of CD8+ T cells in ALS mice increased the number of surviving motoneurons. Moreover, CD8+ T cells expressing the ALS-causing mSOD protein have been described to recognize and selectively kill motoneurons in vitro. To exert their cytotoxic function, CD8+ T cells carrying mSOD1 must be able to recognize neuron antigens inside MHC-I complex at the surface of the motoneurons. Moreover, analysis of the T cell receptor (TCR) diversity supports the evidence that self-reactive CD8+ T lymphocytes infiltrate the CNS of ALS mice and exert cytotoxic functions. Fine-tuning of immune response is usually obtained by multiple regulatory processes, all belonging to the immune tolerance network, that are in place to prevent potentially deleterious immune responses against self-tissues. Regulatory T cells (Treg) are CD4 T lymphocytes characterized by the expression of the Foxp3 transcription factor. This T cell subset controls the immune effector response in terms of clonal expansion, differentiation, cytokine profile and tissue migration and is indispensable for the maintenance of immune self-tolerance [88][48]. Clinical studies in humans and in transgenic mouse models pointed out the role of Tregs in ALS pathophysiology. In humans and in mouse ALS models, Tregs infiltrating the Central Nervous System have been observed to suppress neuroinflammatory processes and promote the activation of neuroprotective microglia. Thus, immune-modulation strategies aimed at increasing Treg number and enhancing its functional effectiveness might be considered relevant to promote neuroprotective activity in ALS [89][49]. mTOR is an evolutionarily conserved serine/threonine protein kinase that directly influences T cell differentiation and proliferation by integrating environmental cues (nutrients, energy stores and growth factors) with immunity functions [90][50]. mTOR activity exerts opposite effects on effector T lymphocytes and on Treg. Indeed, mTOR inhibition strongly favors Treg differentiation and expansion while modulating T cell effector functions [91][51]. Compelling evidence indicate that progressive metabolic conversions, usually mediated by mTOR-dependent pathways, underlie the generation of proper effector functions during T cell response. Recent evidence indicates that SOD1 represents one of the major targets of the mTOR enzyme. Indeed, reversible mTOR-dependent SOD1 phosphorylation has been described to mediate SOD1 inhibition [91,92][51][52]. These observations propose a complex scenario in which SOD1/mTOR intracellular interplay may finely tune T cell activity [93,94][53][54]; instead, no data are available on the role of SOD1 in regulating mTOR dependent pathways. In this context, the recent correlation of increasing SOD1 intracellular levels in T cells with the presence of circulating Tregs in a cohort of subjects affected by Multiple Sclerosis undergoing effective immune-modulating treatment [24][55], strongly supports the idea that a SOD1-mTOR regulatory network [93,94][53][54] may participate in the complex mechanisms regulating immune-modulation processes. Hydrogen peroxide and superoxide anion are involved in TCR-dependent signaling and adaptive immune response activation [23][56]. Indeed, antigen-dependent stimulation of human T lymphocytes can modify SOD1 intracellular localization in T cells, mediating a clear SOD1 recruitment by TCR clusters. In addition, increased SOD1 mRNA production and microvesicle secretion of the enzyme, by activated human T lymphocytes, has been observed [23][56]. In vitro administration of recombinant human SOD1 to activated human T cells increases their IL17 production, a key cytokine in induction/maintenance of chronic inflammatory processes [25,26][57][58]. This effect is mediated by SOD1-dependent enzymatic activity, since SOD1 molecule lacking dismutase activity (Apo SOD1), it is unable to affect T cell cytokine production. Furthermore, hydrogen peroxide addition to activated T-cells mimics the SOD1 effect [24][55]. These data indicate, as summarized in Figure 1, that SOD1 effects on inflammatory process may be, at least partially, linked to its hydrogen peroxide production; this molecule is more stable than other ROS and can freely migrate outside cells and between different cell compartments [24][55].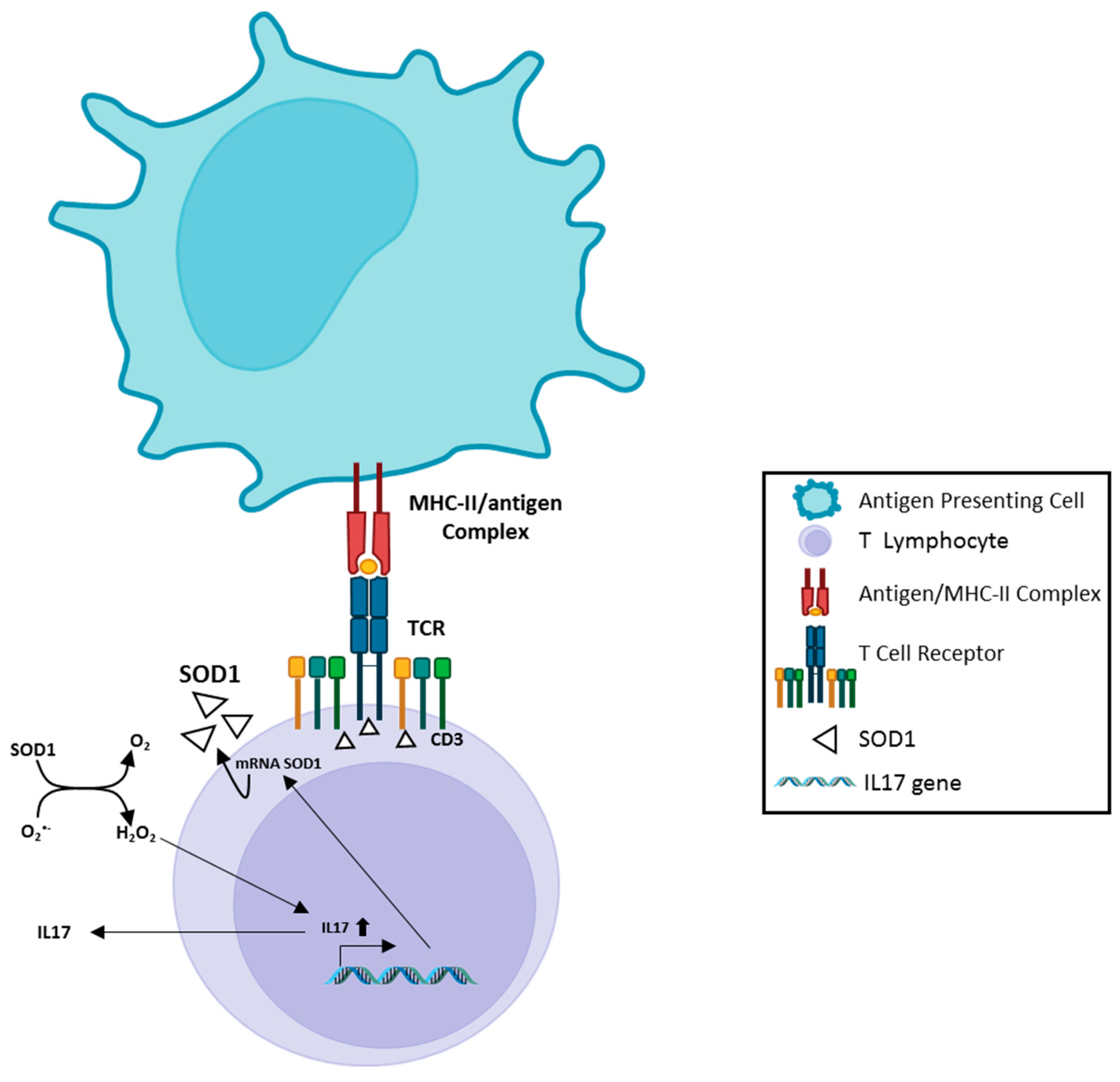
4. SOD1, Muscarinic M1 receptor and Ca2+ level in neurons
Accumulation of Ca2+ in neurons, with consequent activation of Ca-dependent pathways, has been largely associated with cell death [106][59]. Intracellular Ca2+ increase might be mediated by different mechanisms, as represented by several voltage-gate-dependent calcium channels (VGCCS), metabotropic (M1 muscarinic) receptors and ionotropic glutamate-dependent receptors (N-Methyl-D-aspartate (NMDA) and non-NMDA molecules). In addition, the intracellular Ca2+ increase can be mediated by the involvement of metabotropic glutamate receptors (GRM) [107][60].
SOD1 interaction with muscarinic M1 receptor, in human neuroblastoma SK-N-BE cells, can activate ERK1/2 and AKT kinases in a dose/time-dependent manner. This effect was strongly reduced by M1 receptor silencing, as well as by using the M1 antagonist pirenzepine. Moreover, the incubation of SK-N-BE and the neuroblastoma–spinal motoneuron fusion NSC-34 cell line with mutant SOD1G93A significantly increased their intracellular Ca2+ concentration, as compared with wild-type SOD1 treatment [111][61].
Five muscarinic receptor subtypes (m1–m5) have been identified in T lymphocytes [115,116][62][63]; m1, m3 and m5 muscarinic receptor subtypes are coupled to Gq/11, which, upon stimulation, mediate activation of phospholipase C activity, resulting in increases in Ca2+ availability inside cells [117][64]. Moreover, acetylcholine is synthesized and released by T-lymphocytes, acting as an autocrine/paracrine factor, likely involved in immune function regulation [117][64].
These observations add further complexity to the biological scenario involved in the regulation of the neuroinflammatory context.
References
- Atkin, J.D.; Farg, M.A.; Soo, K.Y.; Walker, A.K.; Halloran, M.; Turner, B.J.; Nagley, P.; Horne, M.K. Mutant SOD1 inhibits ER-Golgi transport in amyotrophic lateral sclerosis. J. Neurochem. 2014, 129, 190–204.
- Andrus, P.K.; Fleck, T.J.; Gurney, M.E.; Hall, E.D. Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 1998, 71, 2041–2048.
- Ezzi, S.A.; Urushitani, M.; Julien, J.P. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J. Neurochem. 2007, 102, 170–178.
- Elam, J.S.; Taylor, A.B.; Strange, R.; Antonyuk, S.; Doucette, P.A.; Rodriguez, J.A.; Hasnain, S.S.; Hayward, L.J.; Valentine, J.S.; Yeates, T.O.; et al. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat. Struct. Biol. 2003, 10, 461–467.
- Stathopulos, P.B.; Rumfeldt, J.A.O.; Scholz, G.A.; Irani, R.A.; Frey, H.E.; Hallewell, R.A.; Lepock, J.R.; Meiering, E.M. Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis show enhanced formation of aggregates in vitro. Proc. Natl. Acad. Sci. USA 2003, 100, 7021–7026.
- Rodriguez, J.A.; Valentine, J.S.; Eggers, D.K.; Roe, J.A.; Tiwari, A.; Brown, R.H., Jr.; Hayward, L.J. Familial amyotrophic lateral sclerosis-associated mutations decrease the thermal stability of distinctly metallated species of human copper/zinc superoxide dismutase. J. Biol. Chem. 2002, 277, 15932–15937.
- Bendotti, C.; Carrì, M.T. Lessons from models of SOD1-linked familial ALS. Trends Mol. Med. 2004, 10, 393–400.
- Rakhit, R.; Cunningham, P.; Furtos-Matei, A.; Dahan, S.; Qi, X.F.; Crow, J.P.; Cashman, N.R.; Kondejewski, L.H.; Chakrabartty, A. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J. Biol. Chem. 2002, 277, 47551–47556.
- Alexander, G.M.; Erwin, K.L.; Byers, N.; Deitch, J.S.; Augelli, B.J.; Blankenhorn, E.P.; Heiman-Patterson, T.D. Effect of transgene copy number on survival in the G93A SOD1 transgenic mouse model of ALS. Brain Res. Mol. Brain Res. 2004, 130, 7–15.
- Andersen, P.M.; Nilsson, P.; Ala-Hurula, V.; Keränen, M.L.; Tarvainen, I.; Haltia, T.; Nilsson, L.; Binzer, M.; Forsgren, L.; Marklund, S.L. Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn-superoxide dismutase. Nat. Genet. 1995, 10, 61–66.
- Alexianu, M.E.; Ho, B.K.; Mohamed, A.H.; La Bella, V.; Smith, R.G.; Appel, S.H. The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann. Neurol. 1994, 36, 846–858.
- Beckman, J.S.; Estévez, A.G.; Crow, J.P.; Barbeito, L. Superoxide dismutase and the death of motoneurons in ALS. Trends Neurosci. 2001, 24, S15–S20.
- Howland, D.S.; Liu, J.; She, Y.; Goad, B.; Maragakis, N.J.; Kim, B.; Erickson, J.; Kulik, J.; DeVito, L.; Psaltis, G.; et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc. Natl. Acad. Sci. USA 2002, 99, 1604–1609.
- Beers, D.R.; Henkel, J.S.; Xiao, Q.; Zhao, W.; Wang, J.; Yen, A.A.; Siklos, L.; McKercher, S.R.; Appel, S.H. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16021–16026.
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775.
- Sun, H.; Bénardais, K.; Stanslowsky, N.; Thau-Habermann, N.; Hensel, N.; Huang, D.; Claus, P.; Dengler, R.; Stangel, M.; Petri, S. Therapeutic potential of mesenchymal stromal cells and MSC conditioned medium in Amyotrophic Lateral Sclerosis (ALS)--in vitro evidence from primary motor neuron cultures, NSC-34 cells, astrocytes and microglia. PLoS ONE 2013, 8, e72926.
- Atkin, J.D.; Farg, M.A.; Turner, B.J.; Tomas, D.; Lysaght, J.A.; Nunan, J.; Rembach, A.; Nagley, P.; Beart, P.M.; Cheema, S.S.; et al. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J. Biol. Chem. 2006, 281, 30152–30165.
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40.
- Taylor, J.P.; Brown Jr, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206.
- Maday, S.; Wallace, K.E.; Holzbaur, E.L.F. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 2012, 196, 407–417.
- Rudnick, N.D.; Griffey, C.J.; Guarnieri, P.; Gerbino, V.; Wang, X.; Piersaint, J.A.; Tapia, J.C.; Rich, M.M.; Maniatis, T. Distinct roles for motor neuron autophagy early and late in the SOD1G93A mouse model of ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E8294–E8303.
- McGeer, P.L.; Itagaki, S.; McGeer, E.G. Expression of the histocompatibility glycoprotein HLA-DR in neurological disease. Acta Neuropathol. 1988, 76, 550–557.
- McGeer, P.L.; McGeer, E.G. History of innate immunity in neurodegenerative disorders. Front. Pharmacol. 2011, 2, 77.
- McGeer, P.L.; Itagaki, S.; Tago, H.; McGeer, E.G. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 1987, 79, 195–200.
- Alexianu, M.E.; Kozovska, M.; Appel, S.H. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology 2001, 57, 1282–1289.
- Pramatarova, A.; Laganière, J.; Roussel, J.; Brisebois, K.; Rouleau, G.A. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J. Neurosci. 2001, 21, 3369–3374.
- Qin, L.; Liu, Y.; Wang, T.; Wei, S.J.; Block, M.L.; Wilson, B.; Liu, B.; Hong, J.S. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J. Biol. Chem. 2004, 279, 1415–1421.
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189.
- Marden, J.J.; Harraz, M.M.; Williams, A.J.; Nelson, K.; Luo, M.; Paulson, H.; Engelhardt, J.F. Redox modifier genes in amyotrophic lateral sclerosis in mice. J. Clin. Investig. 2007, 117, 2913–2919.
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723.
- Barber, S.C.; Mead, R.J.; Shaw, P.J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 2006, 1762, 1051–1067.
- Wu, D.C.; Ré, D.B.; Nagai, M.; Ischiropoulos, H.; Przedborski, S. The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12132–12137.
- Harraz, M.M.; Marden, J.J.; Zhou, W.; Zhang, Y.; Williams, A.; Sharov, V.S.; Nelson, K.; Luo, M.; Paulson, H.; Schöneich, C.; et al. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J. Clin. Investig. 2008, 118, 659–670.
- Harraz, M.M.; Park, A.; Abbott, D.; Zhou, W.; Zhang, Y.; Engelhardt, J.F. MKK6 phosphorylation regulates production of superoxide by enhancing Rac GTPase activity. Antioxid. Redox Signal 2007, 9, 1803–1813.
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783.
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441.
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636.
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226.
- Dobson-Stone, C.; Hallupp, M.; Bartley, L.; Shepherd, C.E.; Halliday, G.M.; Schofield, P.R.; Hodges, J.R.; Kwok, J.B. C9ORF72 repeat expansion in clinical and neuropathologic frontotemporal dementia cohorts. Neurology 2012, 79, 995–1001.
- Ono, S.; Hu, J.; Shimizu, N.; Imai, T.; Nakagawa, H. Increased interleukin-6 of skin and serum in amyotrophic lateral sclerosis. J. Neurol. Sci. 2001, 187, 27–34.
- Henkel, J.S.; Beers, D.R.; Wen, S.; Rivera, A.L.; Toennis, K.M.; Appel, J.E.; Zhao, W.; Moore, D.H.; Powell, S.Z.; Appel, S.H. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol. Med. 2013, 5, 64–79.
- Chiu, I.M.; Phatnani, H.; Kuligowski, M.; Tapia, J.C.; Carrasco, M.A.; Zhang, M.; Maniatis, T.; Carroll, M.C. Activation of innate and humoral immunity in the peripheral nervous system of ALS transgenic mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20960–20965.
- Gasco, S.; Zaragoza, P.; García-Redondo, A.; Calvo, A.C.; Osta, R. Inflammatory and non-inflammatory monocytes as novel prognostic biomarkers of survival in SOD1G93A mouse model of Amyotrophic Lateral Sclerosis. PLoS ONE 2017, 12, e0184626.
- Paré, B.; Gros-Louis, F. Potential skin involvement in ALS: Revisiting Charcot’s observation—A review of skin abnormalities in ALS. Rev. Neurosci. 2017, 28, 551–572.
- Nardo, G.; Trolese, M.C.; de Vito, G.; Cecchi, R.; Riva, N.; Dina, G.; Heath, P.R.; Quattrini, A.; Shaw, P.J.; Piazza, V.; et al. Immune response in peripheral axons delays disease progression in SOD1(G93A) mice. J. Neuroinflammation 2016, 13, 261.
- Baruch, K.; Deczkowska, A.; David, E.; Castellano, J.M.; Miller, O.; Kertser, A.; Berkutzki, T.; Barnett-Itzhaki, Z.; Bezalel, D.; Wyss-Coray, T.; et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 2014, 346, 89–93.
- Coque, E.; Salsac, C.; Espinosa-Carrasco, G.; Varga, B.; Degauque, N.; Cadoux, M.; Crabé, R.; Virenque, A.; Soulard, C.; Fierle, J.K.; et al. Cytotoxic CD8(+) T lymphocytes expressing ALS-causing SOD1 mutant selectively trigger death of spinal motoneurons. Proc. Natl. Acad. Sci. USA 2019, 116, 2312–2317.
- Sakaguchi, S.; Ono, M.; Setoguchi, R.; Yagi, H.; Hori, S.; Fehervari, Z.; Shimizu, J.; Takahashi, T.; Nomura, T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol. Rev. 2006, 212, 8–27.
- Sheean, R.K.; McKay, F.C.; Cretney, E.; Bye, C.R.; Perera, N.D.; Tomas, D.; Weston, R.A.; Scheller, K.J.; Djouma, E.; Menon, P.; et al. Association of Regulatory T-Cell Expansion With Progression of Amyotrophic Lateral Sclerosis: A Study of Humans and a Transgenic Mouse Model. JAMA Neurol. 2018, 75, 681–689.
- de Candia, P.; Procaccini, C.; Russo, C.; Lepore, M.T.; Matarese, G. Regulatory T cells as metabolic sensors. Immunity 2022, 55, 1981–1992.
- Procaccini, C.; Matarese, G. Regulatory T cells, mTOR kinase, and metabolic activity. Cell Mol. Life Sci. 2012, 69, 3975–3987.
- Tsang, C.K.; Zheng, X. F S. A balancing act: mTOR integrates nutrient signals to regulate redox-dependent growth and survival through SOD1. Mol. Cell Oncol. 2018, 5, e1488372.
- Tsang, C.K.; Chen, M.; Cheng, X.; Qi, Y.; Chen, Y.; Das, I.; Li, X.; Vallat, B.; Fu, L.W.; Qian, C.N.; et al. SOD1 Phosphorylation by mTORC1 Couples Nutrient Sensing and Redox Regulation. Mol. Cell 2018, 70, 502–515.e508.
- Damiano, S.; Sozio, C.; La Rosa, G.; Guida, B.; Faraonio, R.; Santillo, M.; Mondola, P. Metabolism Regulation and Redox State: Insight into the Role of Superoxide Dismutase 1. Int. J. Mol. Sci. 2020, 21, 6606.
- Rubino, V.; Palatucci, A.T.; La Rosa, G.; Giovazzino, A.; Aruta, F.; Damiano, S.; Carriero, F.; Santillo, M.; Iodice, R.; Mondola, P.; et al. Superoxide Dismutase-1 Intracellular Content in T Lymphocytes Associates with Increased Regulatory T Cell Level in Multiple Sclerosis Subjects Undergoing Immune-Modulating Treatment. Antioxidants 2021, 10, 1940.
- Terrazzano, G.; Rubino, V.; Damiano, S.; Sasso, A.; Petrozziello, T.; Ucci, V.; Palatucci, A.T.; Giovazzino, A.; Santillo, M.; De Felice, B.; et al. T cell activation induces CuZn superoxide dismutase (SOD)-1 intracellular re-localization, production and secretion. Biochim. Biophys. Acta 2014, 1843, 265–274.
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59.
- Jin, M.; Akgün, K.; Ziemssen, T.; Kipp, M.; Günther, R.; Hermann, A. Interleukin-17 and Th17 Lymphocytes Directly Impair Motoneuron Survival of Wildtype and FUS-ALS Mutant Human iPSCs. Int. J. Mol. Sci. 2021, 22, 8042.
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal 2013, 18, 522–555.
- Luo, T.; Wu, W.H.; Chen, B.S. NMDA receptor signaling: Death or survival? Front. Biol. 2011, 6, 468–476.
- Zhou, J.; Yi, J.; Fu, R.; Liu, E.; Siddique, T.; Ríos, E.; Deng, H.-X. Hyperactive intracellular calcium signaling associated with localized mitochondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J. Biol. Chem. 2010, 285, 705–712.
- Kawashima, K.; Fujii, T. Extraneuronal cholinergic system in lymphocytes. Pharmacol. Ther. 2000, 86, 29–48.
- Strong, M.J.; Kesavapany, S.; Pant, H.C. The pathobiology of amyotrophic lateral sclerosis: A proteinopathy? J. Neuropathol. Exp. Neurol. 2005, 64, 649–664.
- Dion, P.A.; Daoud, H.; Rouleau, G.A. Genetics of motor neuron disorders: New insights into pathogenic mechanisms. Nat. Rev. Genet. 2009, 10, 769–782.