Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ilona Sadok and Version 2 by Rita Xu.
Tryptophan metabolism plays an essential role in human health. In mammals, about 95% of dietary tryptophan is metabolized through the kynurenine pathway, which is associated with the development of several pathologies, including neurodegeneration. Some of the kynurenine pathway metabolites are agonists of the aryl hydrocarbon receptor involved in metabolic functions, inflammation, and carcinogenesis.
- tryptophan metabolites
- kynurenine pathway
- kynurenine
1. Introduction
Tryptophan (TRP) is an exogenous amino acid that cannot be synthesized in the human body. It must be delivered through nutritional sources and, in the organism, it is found bound to albumin or free form [1]. In the human organism, TRP is involved in the biosynthesis of protein and regulation of metabolic networks as it is a precursor for important biologically active compounds like coenzymes nicotinamide adenine dinucleotide (NAD+), and NAD phosphate (NADP+), serotonin, tryptamine, melatonin, niacin, kynurenine (KYN), and indole and indolic acid derivatives [2][3][2,3]. Its concentration in the body is lower than that of other amino acids and it might play a rate-limiting role in protein synthesis [4]. The gut microbiota utilizes ~4–6% of TRP for the production of indole, indican, tryptamine, skatole, and indole acid derivatives [5]. In the mammal’s brain, TRP is metabolized to serotonin (5-hydroxytryptamine)—a neurotransmitter that modulates neural activity and a wide range of neuropsychological processes like mood, perception, reward, anger, aggression, appetite, memory, sexuality, and attention [6]. In the pineal gland, serotonin serves as a precursor for the synthesis of melatonin, which is involved in the regulation of circadian rhythm, and the reproductive and immune systems [7]. Approximately, 3% of dietary TRP is utilized for serotonin synthesis throughout the body (1% is utilized in the brain) [8]. Alternatively, TRP might be converted to a trace amount of tryptamine—an important neuromodulator of serotonin [3]. About 95% of intake TRP is metabolized via the kynurenine pathway (KP), producing KYN and other derivatives [9]. KYN secretion by different cells plays an important role in immune privilege during infections, inflammations, pregnancy, and cancer. The contributions of the different metabolic pathways of TRP utilization may differ under physiological and pathological statuses.
Both excessive intake and deficiency of TRP can unbalance its homeostasis and affect human health [4]. The impact of TRP supplementation on human health has so far been studied mainly in terms of serotonin pathway activation. The benefits of TRP loading on human cognition, mood, and sleep as a result of serotonergic stimulation have been widely reported [10]. The KP is the main metabolic route of this amino acid. Moreover, food might contain KP metabolites itself, which has been confirmed by many studies [11][12][13][14][15][16][17][18][19][11,12,13,14,15,16,17,18,19]. Some of them are essential for the proper functioning of the body and have a beneficial effect, e.g., kynurenic acid (KYNA) or nicotinamideadenine dinucleotide. On the other hand, KP delivers some metabolites which could exert cytotoxic (e.g., KYN, 3-hydroxyanthranilic acid (3HAA), and 3-hydroxykynurenine (3HKyn)) and neurotoxic (e.g., quinolinic acid (QA)) impacts. Notably, elevated levels of some KP metabolites are associated with various diseases, e.g., increased KYN and KYNA are connected with inflammatory bowel disease [20] and ulcerative colitis [21], respectively. Providing compounds from KP with food can positively or negatively affect human health.
2. Kynurenine Pathway—An Overview
In mammals, KP (Figure 1) is initiated through the activity of the enzymes: indoleamine-2,3-dioxygenases (IDO1 and IDO2) or tryptophan-2,3-dioxygenase (TDO). TDO acts mainly in the liver, while IDO has a wide tissue and cellular distribution. As a result of this reaction, N-formylkynurenine (NFK) is formed which is instantaneously converted by NFK formamidase to L-KYN—the first stable KP metabolite [22][27]. Degradation of L-KYN proceeds in three ways: (1) enzymes family of kynurenine aminotransferase (KAT) leads to the production of KYNA; (2) L-KYN can be hydrolyzed by kynureninase to anthranilic acid (AA); and (3) kynurenine mono-oxygenase (KMO) can lead to hydroxylation of L-KYN resulting in 3HKyn formation. The activity of some KP enzymes (IDO and KMO) is also upregulated by immune stimulation [23][28]. Further transformation of 3HKyn is accomplished by kynureninase and causes 3HAA formation [24][29]. The next product, 2-amino-3-carboxymuconic 6-semialdehyde (ACMS), is formed as a result of 3-hydroxyanthranilate-3,4-dioxygenase action. This unstable metabolite can be converted in two different ways: (1) preferably via QA, which, subsequently, undergoes the transformation and leads to the production of the essential redox cofactor nicotinamideadenine dinucleotide (NAD+) and (2) less efficiently 2-amino-3-muconic acid-6-semialdehyde (AMS), which, subsequently, generates picolinic acid (PIC) resulting from non-enzymatic cyclization or it is metabolized to acetyl CoA [22][25][27,30]. Alternatively, the transamination of 3HKyn by KAT leads to xanthurenic acid (XA) formation, whereas the dimerization of 3HAA produces cinnabarinic acid (CA) [23][28].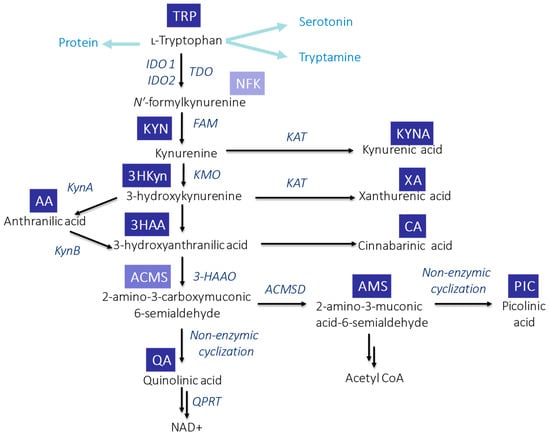
Figure 1. Schematic overview of TRP metabolism via KP in mammals. Abbreviations: ACMSD—2-amino-3-carboxymuconic acid semialdehyde decarboxylase; FAM—N′-formylkynurenineformamidase; 3-HAAO—3-hydroxyanthranilic acid 3,4-dioxygenase; IDO1—indoleamine-2,3-dioxygenase 1; IDO1—indoleamine-2,3-dioxygenase 2; KAT—kynurenine aminotransferase; KMO—kynurenine mono-oxygenase; KynA—kynureninase A; KynB—kynureninase B; QPR—quinolinate phosphoribosyl transferase; and TDO—tryptophan-2,3-dioxygenase.
3. Circulation of KP Metabolites
In the human body, approximately <1% of ingested TRP is utilized for protein synthesis. TRP is transported by large neutral amino acid transporters mainly into the gut, where it is metabolized by microbiota [5]. The rest enters portal circulation and undergoes liver metabolism [26][31]. Notably, about 75–95% of circulating TRP is bound to albumin, but only free TRP can cross the blood–brain barrier (BBB). The unbounded TRP can be further metabolized along four degradation pathways [3][26][3,31]. Gut microbiota modulates intestinal TRP metabolism. Commensal microbes can transform TRP into tryptamine (by tryptophan decarboxylases), indole and its derivatives (by tryptophanase), and serotonin (by tryptophan synthetase). However, the initiation of TRP degradation via KP occurs by the activation of Toll-like receptors (TLRs) by microbial components. Notably, butyrate—produced by gut microbes—suppresses KYN production by downregulation of intestinal IDO expression via various mechanisms [27][32]. More than 60% of KYN is directly transported from peripheral circulation to the brain, where it is transformed into other neuroactive compounds [28][33]. 3HKyn and AA also have the ability to cross BBB, while KYNA, 3HAA, and QA cross it very poorly [28][29][30][33,34,35]. Furthermore, KYN can cross the placenta and fetal blood–brain barrier [31][36]. In peripheral tissues (liver and kidney), phagocytes (monocytes and macrophages), and microglia cells, KMO predominantly break down KYN to 3HKyn, which is further cleavaged to 3HAA and QA, leading to NAD+ formation [23][28]. Alternatively, KYN can be hydrolyzed to AA—a precursor for QA production. In astrocytes, however, KAT is catalyzed to KYN and converted to KYNA. Furthermore, glial cells and neurons produce PIC [23][28]. After oral ingestion, KYN is absorbed into the intestine [28][33]. Also, KYNA can be easily absorbed from the lumen of the digestive system into the bloodstream and transported to the liver and the kidneys [32][33][37,38]. In blood, its concentration achieves the highest level 15–30 min after ingestion, and back to the basal level after 2 h [33][38]. After ingestion, a successive increase in KYNA level in bile, pancreatic juice, and intestinal lumen is observed [34][39]. Under normal conditions, the majority of KP metabolites are excreted in the urine [34][35][36][39,40,41]. The presence of some KP metabolites in faeces and sweat was also demonstrated [34][36][39,41].4. Biological Functions of KP Metabolites
The role of KP is connected with many diseases and conditions. The metabolism of TRP is related to neuropsychiatric and cardiovascular diseases, inflammation, and cancer, and KP metabolites have various biological functions (Table 1).Table 1. Comparison of positive and negative health effects of the KP metabolites.
| Biological Effect | KP Metabolite | Ref. |
|---|---|---|
| Anticonvulsant properties | KYNA, PIC | [23][28] |
| Anti-inflammatory properties | 3HAA, 3HKyn | [23][37][28,42] |
| Antimicrobial activity | KYN, KYNA, CA, PIC | [23][38][28,43] |
| Antioxidant properties | KYNA, XA, AA, 3HKyn | [23][37][39][28,42,44] |
| Antiviral properties | PIC | [23][28] |
| Immunomodulation | KYN, CA | [23][38][28,43] |
| Lipid peroxidation | QA | [40][45] |
| Neurotoxicity | 3HKyn, QA | [37][40][42,45] |
| Neuroprotective properties | KYNA | [23][28] |
| Oxidative stress regulation | KYN, 3HKyn, 3HAA | [23][28] |
| Proconvulsant properties | QA | [23][28] |
| Pro-oxidant properties | 3HKyn, QA | [23][37][28,42] |
| Reduction of lipid peroxidation | KYNA | [23][28] |
| Transcription factor | KYN | [23][28] |
| Vasodilator in endothelial cells | KYN | [23][28] |