Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Elias Kouroumalis and Version 2 by Rita Xu.
The pathogenesis of inflammatory bowel disease (IBD) implicates several interconnecting factors. Immunity and external factors interact, and most aspects are still under investigation. Autophagy and apoptosis are two critical pathways that decide the fate of the individual cells of the intestinal mucosa.
- inflammatory bowel disease
- apoptosis
- autophagy
1. Introduction
Inflammatory bowel disease (IBD) is an intestinal inflammatory disease that includes Crohn’s disease (CD) and ulcerative colitis (UC). CD is a chronic granulomatous lesion that can affect the entire gastrointestinal tract but usually involves the colon and small intestine. UC is a chronic, non-specific inflammation that affects the colonic mucosa and submucosa [1][2][1,2]. The small intestinal epithelium is comprised of villi and the crypts of Lieberkühn, while the colonic epithelium consists only of crypts. Inflammation of the intestinal mucosa in association with villus atrophy and a reduction in crypts are histological characteristics of IBD [3].
Globally, more than 2.5 million people of European origin have the disease [4]. However, increasing numbers of IBD cases are observed in Asia and Africa, where it was practically absent at the beginning of this century [5]. The pathogenesis of IBD is a multi-factorial process implicating immunological, environmental, genetic and microbial factors [6]. The intestinal mucosal barrier is the first defense against harmful invaders from the bowel lumen. This barrier consists of three layers, namely the surface mucus, the epithelial cell layer, and the cells of the immune system of the submucosa [7]. The surface mucus inhibits the direct contact of the intestinal bacteria with the epithelial cells [8]. The intestinal epithelial cells consist of enterocytes, goblet cells, Paneth cells, tuft cells and endocrine cells. They are the site of absorption and production of mucus and several important proteins [9]. The immune cells of the submucosa are neutrophils, lymphocytes and macrophages fighting the pathogens that can penetrate into the submucosa. They are involved in the handling of antigens and the secretion of multiple cytokines [10][11][10,11].
Recently, the role of two important cellular pathways has been studied in connection with the pathogenesis of IBD. Autophagy and apoptosis are two mechanisms that actively participate in the integrity of the intestinal mucosa [12].
Autophagy is a key process for cell survival and tissue homeostasis, turning damaged organelles and cellular waste into useful reusable metabolites [13][14][13,14]. Starvation and infection may initiate autophagy and increase cell viability. Autophagy has been identified in both the small intestinal and colonic mucosa, and it may be implicated in the pathophysiology of IBD [13][14][13,14]. On the contrary, apoptosis eliminates damaged cells [15]. The balance between autophagy and apoptosis is one of the factors that determine the fate of intestinal cells, although other forms of cellular death, such as necroptosis or pyroptosis, can also lead to cellular death in IBD in response to inflammatory signals [13].
The complex nature of autophagy and its interplay with apoptosis make it difficult to delineate their exact role in IBD, although current evidence has indicated the participation of autophagy in mucosal homeostasis [16]. Thus, autophagy is involved in the secretion of pro-inflammatory cytokines and activation of inflammasomes, while reactive oxygen species (ROS) are increased in autophagy-deficient macrophages, impairing submucosal immunity [17]. Autophagy is also involved in several functions of intestinal epithelial cells (IECs) and goblet cells, particularly during infection or ER stress [18]. An example is the Salmonella infection, where a deficiency of autophagy in IECs leads to bacterial dissemination and increased inflammation [18]. The intestinal epithelium may then become vulnerable to the infiltrating microbes and their products, and its fate will depend on the interaction of autophagy with the various forms of cellular death [19]. Autophagy is also important for the function of Paneth cells. Paneth cells secrete antimicrobial peptides (AMPs) and other peptides, including lysozymes, alpha-defensins and phospholipase A1, when invaders stimulate the crypt lumen [20]. Paneth cells are critical in the maintenance of a normal microbiota. A form of autophagy to remove pathogens named xenophagy is operative in Paneth cells, allowing maintenance of the normal metabolic functions of the host [21]. Dysfunction of Paneth cells leads to reduced secretion of AMPs and dysbiosis, which is a dysregulation of the composition of intestinal microbiota. Dysbiosis may be implicated in the pathogenesis of IBD [22].
2. An Overview of Autophagy and Apoptosis
2.1. Autophagy
Autophagy is a crucial degradative process that is vital for cellular economy and homeostasis. Proteins, lipids, damaged organelles and pathogens are degraded and the products re-used for the synthesis of cellular constituents [23]. Autophagy research led to two Nobel Prizes, one to Christian De Duve, who described the significance of lysosomes in biology, and the second to Yoshinori Ohsumi, who clarified the mechanisms of autophagy [24]. In reality, the process of autophagy is a series of phosphorylations and de-phosphorylations. Three kinases are the main regulators of autophagy: the mammalian target of rapamycin (mTOR), Unc-51-like autophagy activating kinase (ULK1) and the AMP-dependent protein kinase (AMPK) [25]. Decreased nutrients and ATP levels and increased levels of reactive oxygen species (ROS) and damaged DNA are the initiators of AMPK and mTOR [26]. Phosphorylation of ULK1 by mTOR reduces its activity and decreases autophagy, while phosphorylation by AMPK at a different site activates ULK1 and autophagy. AMPK inhibits mTOR, leading to the highest activation of ULK1 [27]. Activated ULK1 phosphorylates Beclin1 and induces autophagy [28][29][28,29]. At the same time, JNK-1 phosphorylates Bcl-2, leading to its dissociation from Beclin1. Free Bcl-2 inhibits apoptosis through binding to BAX and BAK proteins. Free Beclin1 subsequently binds to Vps34–Vps15 to promote autophagy through the formation of the class III PI3K complex consisting of Vps34–Vps15–Beclin1 [30][31][30,31]. The next step is the autophagosome formation, which is the direct result of ULK1 phosphorylation. Here, the gatekeeper is the phosphorylation of autophagy-related protein 13 (Atg13), leading to a complex formation with Atg5–Atg12, Atg16L1 and LC3-II. Additional proteins that are often studied in autophagy research and are associated with autophagosomes include LC3 (Atg8) and protein sequestosome 1 (p62 SQSTM1) [32][33][32,33]. Finally, autophagosomes fuse with lysosomes, forming autolysosomes during the so-called autophagic flux. When autophagosomes are produced faster than autophagic flux, or when the flux is repressed, the levels of LC3 and p62 will increase [34]. Constituents that are destined for lysosomal degradation are either labeled by ubiquitin or attached to cargo receptors such as SQSTM1, and calcium-binding and coiled-coil domain-containing protein 2 (CALCOCO2). The lysosomal contents recirculate and useful elements are used again [23]. Another important regulator of autophagy is transcription factor EB (TFEB), which is a controller of lysosomal biogenesis genes [35] in association with TFE3 (transcription factor binding to IGHM enhancer 3) [36]. Reduced expression of TFEB leads to the exacerbation of inflammation [37]. mTOR activation decreases TFEB activity, and the autophagic machinery is suppressed [38].2.2. Mitophagy
Mitophagy is a specialized form of protective autophagy that selectively degrades damaged mitochondria irrespective of the cause of the damage [39][40][39,40]. Two signal pathways initiate mitophagy, either through the PINK1 (PTEN-induced putative kinase 1)–PARKIN (parkin RBR E3 ubiquitin protein ligase) system or through a PINK1/PARKIN-independent pathway [41][42][41,42]. Normal mitochondria accumulate PINK1 in the inner mitochondrial membrane (IMM) assisted by the TOMM (translocase of the outer mitochondrial membrane) and TIMM23 (translocase of inner mitochondrial membrane 23) proteins. PINK1 is further cleaved by PARL (presenilin-associated rhomboid like). Damaged mitochondria, on the other hand, accumulate PINK1 in the outer mitochondrial membrane (OMM) instead of the IMM [43][44][43,44], leading to the recruitment of ubiquitin and PARKIN from the cytoplasm to the OMM [45]. PARKIN links phosphoubiquitin molecules to several proteins of the mitochondrial surface, such as MFN1 (mitofusin 1) and MFN2 [42][46][47][42,46,47]. Cargo receptor proteins such as SQSTM1/p62 (sequestosome 1), OPTN (optineurin) and CALCOCO2, among others, bind to these OMM proteins to start autophagosome formation [47][48][47,48]. Mitophagy may be upregulated by phosphorylation of OPTN via the activation of TBK1 (TANK-binding kinase 1) [49][50][49,50]. Details of the mitophagy mechanisms have been recently published [51][52][51,52].2.3. Apoptosis
Apoptosis is a form of programmed cell death. During apoptosis, cellular elements are not spilled into the surrounding environment and do not induce inflammation. Apoptosis, in fact, is a downstream activation of a series of caspases. In the caspase chain, each inactive enzyme pro-form is cleaved and activated by the previous active caspase [53]. There are two apoptotic pathways, namely the intrinsic and the extrinsic pathways [54]. The intrinsic pathway starts with the activation of the pro-apoptotic BH3 domain proteins such as BAX and BAK, which drill pores in the OMM, leading to the reduction of ATP and the liberation of cytochrome c and apoptosis-inducing factor (AIF). Cytosolic AIF is translocated into the nucleus and causes DNA fragmentation. Cytochrome c with Apaf-1 (apoptotic protease-activating factor-1) induces the activation of pro-caspase 9, leading to the activation of pro-caspases 3 and 7, which execute the cell [55]. The extrinsic apoptosis pathway is induced by activation of death receptors such as FAS, DR4/DR5 and TNFR1 by their ligands in the cell surface (FAS ligand, TNF-related apoptosis-inducing ligand (TRAIL) and TNFα, respectively). After death receptor activation, the protein FADD associates with the receptors and causes the activation of pro-caspase 8. The death receptor—FADD—pro-caspase 8 complex is called the death-inducing signaling complex (DISC). The anti-apoptotic protein FLICE-like inhibitor protein (FLIP) inhibits the activation of caspase 8 [56]. Caspase 8 may lead to cell death in one of two ways The first is through the activation of the pro-apoptotic BH3 domain protein BID, which inhibits anti-apoptotic BH3 domain proteins such as BCL-2, BCL-XL and MCL1, allowing the lethal BAX and BAK proteins to aggregate in the OMM and form pores [57] in a similar way to the activation of the intrinsic pathway. The second way for caspase 8 is to directly activate pro-caspase 3, omitting the mitochondrial step [57]. One of the most important inducers of apoptosis through the intrinsic pathway is endoplasmic reticulum (ER) stress. Under normal conditions, the three main ER stress transducers (activating transcription factor 6 (ATF6), the inositol-requiring enzyme 1 (IRE1) and the PRKR-like endoplasmic reticulum kinase (PERK) are inactive and attached to BiP (immunoglobulin heavy-chain binding protein, also referred to as glucose-regulated protein GRP 78). When unfolded proteins accumulate into the ER lumen, BiP dissociates from the ER stress transducers. IRE1 and PERK are involved in apoptosis. IRE1 activates JNK and stimulates the phosphorylation of Jun. Active PERK phosphorylates eukaryotic initiation factor 2α (eIF2α), which results in the successive downregulation of protein translation. eIF2α also induces preferential translation of transcription factor ATF4. Thereby, ATF4 induces expression of C/EBP-homologous protein (CHOP) and ATF3. These proteins trigger an apoptotic program [58]. The most important inducer of apoptosis through the extrinsic pathway is tumor necrosis factor α (TNFα). Under normal circumstances, complex I is formed when RIPK1 (receptor-interacting protein kinase 1) leads to the activation of the pro-inflammatory and protective pathway NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells). In pathologic conditions, either complex IIa or complex IIb can be formed, leading to the activation of caspase-8, which in complex IIa cleaves receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and downstream pro-caspases 3 and 7 to initiate apoptosis. Complex IIb, on the other hand, depends on the active intact RIPK1 kinase to activate caspase-8 and apoptosis [59].2.4. Ferroptosis
Ferroptosis is a recently described regulated cell death induced by iron accumulation, lipid peroxidation, and the production of ROS, depending on the activity of NADPH oxidases [60]. Cells defend themselves from the lethal lipid peroxidation and ROS formation through different anti-oxidative systems. The most important is the Xc-antiporter system consisting of the SLC7A11 and SLC3A2 subunits, which imports cysteine turning into cysteine and exports glutamine turning into glutamate. γ-glutamylcysteine is formed, followed by the production of glutathione (GSH) and glutathione peroxidase (GPX4) to neutralize the ROS and lipid peroxides produced from either polyunsaturated fatty acids (PUFAs) or phospholipids of the cell membrane through the action of lipoxygenases (LOXs). GSH formation requires nicotinamide adenine dinucleotide phosphate (NADPH). Long-chain-fatty-acid-CoA ligase 4 (ACSL4) facilitates the formation of phosphatidylethanolamine (PE)-PUFAs, finally turning into PE-PUFA hydroperoxides (OOH). Downregulation of GPX4 production favors lipid peroxidation and damage to lipid membranes and mitochondria [61][62][61,62]. In addition to the enzymatic oxidation of PUFAs by LOXs, a nonenzymatic reaction, such as a Fenton-type reaction, can also oxidize PUFAs [63][64][63,64]. P53 enhances the activity of LOX and promotes lipid peroxidation [62]. However, p53 has a dual role in the regulation of ferroptosis. p53 may induce ferroptosis through inhibition of SLC7A11 or upregulation of arachidonate lipoxygenase (ALOX). On the other hand, p53 may inhibit ferroptosis through either inhibiting dipep-tidyl-peptidase-4, (DPP4) activity or activating the cyclin-dependent kinase inhibitor 1A (CDKN1A)/p21 pathway [65]. Ferrous iron (Fe2+), through the Fenton reaction, promotes the conversion of PE-PUFA into PE-PUFA-OOH, leading to ferroptosis [66]. Besides the Xc system, there are two more antioxidant systems counteracting ferroptosis, namely the ferroptosis suppressor protein 1–coenzyme Q10-NADPH (FSP1–CoQ10-NADPH) axis and the dihydroorotate dehydrogenase (DHODH)–CoQ-NADPH axis acting in parallel to the GPX4-GSH axis [67]. The three main antioxidant systems have discrete cellular localization: the GSH/GPX4 pathway is cytoplasmic, the FSP1–CoQ-NADPH system is located in the cell membrane, and the DHODH–CoQ-NADPH pathway is related to mitochondria. Mitochondria have a critical role in lipid peroxidation and ferroptosis [68][69][68,69]. Deprivation of glucose impairs the tricarboxylic acid cycle (TCA) cycle in mitochondria and inhibits ferroptosis via AMPK kinase signaling [70]. On the other hand, free iron activates the mitochondrial membrane protein mitoferrin 2, which in turn increases iron influx into the mitochondria, promoting ROS production and ferroptosis. Specific inhibitors of ferroptosis, such as deferoxamine (DFO), ferrostatin-1 (Fer-1) and liproxstatin-1 (Lip-1), and potent inducers, such as erastin, RAS-selective lethal 3 (RSL3), FIN56, FINO2, sulfasalazine and sorafenib, have been described [71][72][71,72]. Erastin and FIN 56 deplete GSH and allow for increased levels of lipid peroxides and ferroptosis, while RSL3 and FINO2 directly inhibit GPX4. The ferroptosis inhibitors also antagonize ferroptosis through various mechanisms. Thus, iron chelators reduce the labile free iron, downregulating lipid peroxidation. Fer-1 and Lip-1 protect lipids from autoxidation. Lipoxygenase (LOX) inhibitors, such as zileuton, baicalein and NDGA, could counteract LOXs-induced lipid peroxidation. Thiazolidinedione, an ACSL4 (acyl-CoA synthetase long-chain family member 4) inhibitor, represses the esterification of PUFAs, interfering in the step before the production of PE-PUFA-OOHs [67].2.5. Interaction of Autophagy and Apoptosis
The interplay between apoptosis and autophagy is complex since autophagy is usually an adaptation leading to cell survival and, in most instances, it represses apoptosis. Similar stimuli can initiate either apoptosis or autophagy. However, under certain circumstances, autophagy may turn into an alternative pathway to cellular death, the so-called autophagic cell death. Usually, autophagy and apoptosis move in a mutually exclusive manner. Autophagy and apoptosis may also move in the same direction [73]. Earlier studies demonstrated that TNFα-induced apoptosis was reduced by autophagy [74]. Similar findings were reported in immortal epithelial cells, where autophagy de-graded the death receptor associated protein FADD [75]. The first observation of an inter-action of autophagy and apoptosis was the effect of the anti-apoptotic protein Bcl-2 on the autophagic protein Beclin1, as mentioned before. Beclin1 interacts with Bcl-2 family proteins and dissociation of the complex frees Beclin1 to further promote autophagosome formation [76]. It should be noted that two distinct cellular pools of Bcl-2 exist in each cell. The ER pool is bound to Beclin1, where it inhibits autophagy, and the mitochondrial pool, which is bound to BAX and BAK, where it inhibits apoptosis [77][78][77,78]. A BH3-only protein, either BIK, BAD, or NOXA, can bind to Bcl-2, liberating Beclin1, a process augmented through the phosphorylation of Bcl-2 by c-Jun N-terminal kinase (JNK1) during nutrient deprivation [79][80][81][79,80,81]. Contrary to expectations that the binding of Beclin1 to Bcl-2 would induce apoptosis through neutralization of the anti-apoptotic function of Bcl-2, this is not the case and apoptosis is not induced [82]. The caspase 8 inhibitor FLIP also inhibits autophagy by interfering with Atg3 binding in LC3 lipidation [83]. Different regions within the FLIP protein control its anti-autophagic and anti-apoptotic activities [83], and different pools of FLIP may provide separate regulation of FLIP-mediated autophagy and apoptosis. The autophagy-associated Atg5 protein is also implicated in the modulation of autophagy and apoptosis. During apoptosis, Atg5 is cleaved by active caspase 3 and an amino-terminal molecule is produced [84]. This molecule induces apoptotic cell death, as it inhibits the anti-apoptotic Bcl-2 molecules, allowing BAX and BAK to initiate mitochondrial-induced apoptosis. At the same time, the cleaved Atg5 is not able to promote autophagy [84]. During protracted exposure to apoptotic stimuli, cleavage of Beclin1 by caspase 3 produces two molecules: an N-terminal and a C-terminal that are unable to induce autophagy any more. Moreover, the C-terminal molecule enters the mitochondria and in-creases the sensitivity of cells to apoptotic signals [85]. In contrast to the downregulation of autophagy caused by the cleavage of Atg5 or the similar cleavage of Beclin1, the cleavage of Atg4D by caspase 3 generates a molecule that increases autophagy. Moreover, autophagy inhibits apoptosis via either lysosomal degradation of active caspase-8 or via Beclin1 inhibition of the BH3-only protein BID that activates BAX and BAK [30]. Finally, autophagy and apoptosis may cause cell death when acting in concert. The exact mechanisms are still under investigation. One such mechanism may be the liberation of cathepsins from autolysosomes, which may kill the cell during autophagy and, at the same time, cleave BID, increasing apoptosis via activation of BAX and BAK [86][87][86,87]. Figure 1 shows the complicated interactions of autophagy and apoptosis in connection to ferroptosis.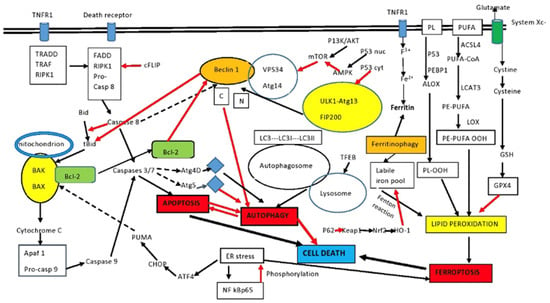
Figure 1. Intersection between apoptosis and autophagy. Autophagy and apoptosis have similar signaling pathways and usually but not always exhibit mutual inhibition. Beclin1 and Bcl-2 are important elements in the interplay between the two. Sustained apoptotic stimuli lead to caspase cleavage of Beclin1 and the C-terminal fragment inhibits autophagy. Although apoptosis-associated cleavage of Beclin1 and Atg5 inhibits autophagy, the cleavage of Atg4D by caspase 3 generates a fragment that increases autophagy. Moreover, autophagy inhibits apoptosis by degrading active caspase 8 or preventing activation of Bid by Beclin1. The effect of P53 on autophagy depends on localization. Cytoplasmic P53 inhibits autophagy, while nuclear P53 activates AMPK and increases autophagy. PUFA and PL of the membrane are the sources of lipid peroxides in ferroptosis. p53 and PEBP1 enhance the activity of LOX to promote lipid peroxidation, resulting in ferroptosis. In addition, LOX promotes the conversion of PE-PUFA into PE-PUFA-OOH, leading to ferroptosis, while the free iron pool promotes lipid peroxidation and ferroptosis through the Fenton reaction. The p62/keap1/Nrf2/HO-1 system decreases ferroptosis, along with the main antioxidant system Xc-. Ferritinophagy is a major source of the labile iron pool. Certain pathways have been omitted for clarity. See the text for details. ACSL4: acyl-CoA synthetase long-chain family member 4; ALOX: arachidonate lipoxygenase; Apaf1: apoptosis signal-regulating kinase; ATF4: activating transcription factor 4; GSH: glutathione; BAK: Bcl-2 homologous antagonist killer; BAX: Bcl-2-associated X protein; Bcl-2: B-cell lymphoma-2; Bid: BH3-interacting-domain death agonist 2; CHOP: CCAAT-enhancer-binding protein homologous protein; FADD: Fas-associating protein with death domain; GPX4: glutathione peroxidase 4; LOX: lipoxygenase; LPCAT3: lysophosphatidylcholine acyltransferase 3; mTOR: mammalian target of rapamycin; PEBP1: phosphatidylethanolamine-binding protein-1; PL: phospholipid; PLOOH: phospholipid hydroperoxides; PE: phosphatidylethanolamine; PUFAs: polyunsaturated fatty acids; PUFA-OOH: polyunsaturated fatty acid containing-phospholipid hydroperoxides; PUMA: p53 upregulated modulator of apoptosis; RIPK1: receptor interacting serine/threonine kinase 1; black arrows: activation; red arrows: inhibition; intermittent arrows: cleavage.